


 下载:
下载:
-
开放科学(资源服务)标识码(OSID):

-
作为我国重点管控的新污染物之一,微塑料近年来引起了全球范围内的广泛关注。微塑料是指粒径小于5 mm的塑料颗粒,在天然水体中被频繁且广泛检出[1-2]。环境因素,比如风吹、日晒等加速了微塑料的老化、分解和破裂,形成大量粒径小于1 000 nm的纳米塑料颗粒[3]。已有研究表明,湖泊中微塑料的颗粒浓度可达900~10 120 p/m3,根据微塑料的来源和降解过程推断,水环境中纳米塑料的浓度可达微米塑料的1014倍[4-5],污染更为普遍。与微米塑料相比,纳米塑料粒径和密度较小,可分散于水体中形成纳米塑料胶体体系,易被多种水生生物误食,导致其生长受限、生殖功能障碍,甚至诱发死亡[6-7]。此外,纳米塑料胶体颗粒具有比表面积大、表面官能团较多和疏水性强等特点,容易与水体中共存的各种天然有机质、营养元素(如Na+和Ca2+)和污染物质(如重金属和有机污染物)发生相互作用,影响纳米塑料胶体的凝聚和分散[8-9]。纳米塑料胶体稳定性的改变一方面会影响塑料颗粒在水体中的沉降、再悬浮和水平迁移等环境行为[1, 10],另一方面,还可能对其表面吸附的营养物质和污染物质在水环境中的迁移和归趋产生影响,从而带来潜在的联合暴露生态风险。因此,水体中纳米塑料胶体稳定性的影响因素及其微观机制成为近期研究热点。
值得注意的是,全球城镇化快速发展和工农业废水的处置不当等使得大量氮和磷等营养物质随排水系统进入环境水体,加剧了天然水体的富营养化[11-13]。水体富营养化促使藻类等浮游植物的过量繁殖,暴发有害藻华[14]。在近40年间,全球约8.8%的湖泊暴发了藻华[15],且藻华暴发面积和频率呈现逐年增加的趋势[16]。随着藻华在全球范围内的蔓延,很多微纳塑料污染的天然水体也面临着日益严重的藻华暴发问题,如中国的滇池[17]和太湖[18]。藻华暴发期间,藻类代谢和残体分解将释放大量溶解性藻源胞内有机质(Intracellular algae organic matter,IOM)进入水体[19-20]。这些IOM主要由蛋白质、氨基酸、多糖等大分子组成,与湖库陆源有机组分腐殖酸等存在显著区别[21]。目前,部分学者研究了环境中单一溶解性有机质组分,如蛋白质、腐殖酸(HA)和富里酸(FA)对纳米塑料稳定性的影响及机理 [4, 22-23]。例如,Dong等[22]研究发现HA可能会通过增强颗粒间的空间位阻作用和静电斥力抑制Na+体系下纳米塑料胶体的聚集,通过吸附在PNs表面的HA与Ca2+产生离子架桥作用促进Ca2+体系下PNs的聚集。Liu等[24]却发现在Na+体系下,与胞外聚合物(EPS)相比,HA能更显著地抑制PNs的聚集。这说明天然有机质类型和组分对纳米塑料稳定性的影响可能存在较大差异。鉴于当前研究所购买的溶解性有机质组分大多为纯化学品,其成分和组分复杂程度与实际IOM存在一定差异。因此,目前还缺乏IOM和环境纳米塑料相互作用的系统研究。
鉴于此,本研究拟以环境中普遍存在的PNs为研究对象,通过动态光散射技术探究IOM组分和质量浓度对PNs在水体中分散和凝聚的影响规律,结合理论计算、荧光和红外光谱分析等表征手段揭示IOM对PNs稳定性的影响机制。研究结果有利于科学认识水体富营养化背景下纳米塑料和相关环境污染物的环境归趋,对环境纳米塑料的防治也具有一定理论和实践意义。
全文HTML
-
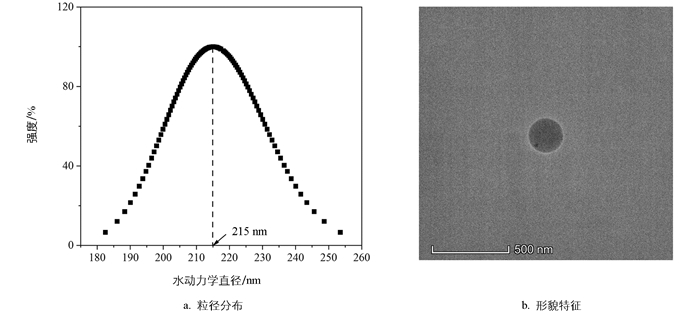
球形的羧基改性PNs颗粒(平均粒径215 nm,图 1)购自天津倍思乐色谱技术开发中心(Tianjin BaseLine Chromtech Research Centre),初始质量浓度为2.5×104 mg/L。实验所需的其他药品,如氯化钠、无水氯化钙、氢氧化钠和盐酸等均为分析纯,购自国药集团化学试剂有限公司。
-
选用购置于中国科学院水生生物研究所淡水藻种库(武汉,中国)的铜绿微囊藻(Microcystis aeruginosa,FACHB-905)进行室内培养并提取实验所需的IOM。在无菌条件下,将铜绿微囊藻批量接种于BG11培养基中,置于恒温智能光照培养箱中培养。培养条件:温度为25±1 ℃,湿度为75% RH,光照强度为3 000 lx,光暗比为14 h/10 h。取培养至对数期—稳定期的藻细胞,利用反复冻融离心法提取藻源IOM[25],利用TOC测定仪测定有机质含量,利用三维荧光光谱技术分析IOM组分。
-
采用时间分辨动态光散射法分别测定NaCl和CaCl2条件下单纯PNs胶体和PNs-IOM混合胶体的凝聚动力学过程。实验所用仪器为NanoBrook Omni多角度粒度与高灵敏Zeta电位分析仪(Brookhaven,美国),测试温度为25 ℃,光源为He-Ne激光光源,入射光波长为640 nm,散射角度为90°。
参照前期相关文献,结合实际水体常见溶解性有机质质量浓度(0~40 mg/L)[26-27],设定本实验的PNs胶体颗粒质量密度为30 mg/L,IOM质量浓度为0、5、20、40 mg/L。实验过程中,在比色皿中依次加入PNs胶体悬液、超纯水、适量的IOM和电解质溶液,确保待测样品总体积为2 mL,体系pH值为6左右,每隔30 s记录一次絮体的动力学粒径数据,连续测定40 min,跟踪凝聚过程中凝聚体粒径随时间的动态变化,凝聚实验完成后,测定各体系的ζ电位。
将凝聚体的粒径增长对凝聚时间求导,得到凝聚速率随时间的变化曲线,并由公式(1)计算胶体凝聚的总体平均凝聚速率[28]:
式中:
$\tilde{v}_{T}\left(f_{0}\right)$ 为t=0时刻到给定时间t=t0(t0>0)时刻的总体平均凝聚速率(nm/min),t0上限为凝聚结束时刻;f0为电解质浓度(mmol/L);D(t)为t时刻胶体颗粒(或凝聚体)的粒径(nm);D0为胶体颗粒的初始粒径(nm)。 -
采用透射电子显微镜(Talos F200X,捷克)表征PNs的形貌,取PNs悬液滴于200目碳涂层铜网中,待格栅网格自然风干10~20 min后上机测定;除测定1.3节胶体凝聚过程中体系的ζ电位外,利用NanoBrook Omni多角度粒度与高灵敏Zeta电位分析仪测定pH值为2~10的PNs、IOM及PNs-IOM的ζ电位,得到胶体的等电点,此时,PNs胶体颗粒质量浓度也设置为30 mg/L,IOM质量浓度为20 mg/L;采用三维荧光光谱仪(FL6500,Perkinelmer,美国)检测IOM、PNs和PNs-IOM凝聚前后体系溶解性有机质的有机组分,其中,激发波长Ex设为200~500 nm,发射波长Em设为250~550 nm,狭缝宽度设为10 nm,光谱扫描速度设为1 200 nm/min;利用傅里叶变换红外光谱仪(Spectrum Two,Perkinelmer,美国)进行凝聚体特定化学基团信息的表征,测定过程中采用全反射模式,光谱范围设定为4 000~400 cm-1,数据间隔为1 cm-1,分辨率为4 cm-1。以上表征中涉及的IOM质量浓度均为20 mg/L。
-
利用经典的DLVO理论计算模型对不同电解质条件下PNs的聚集动力学过程进行拟合,以揭示PNs胶体凝聚的微观机理。根据DLVO理论,纳米塑料颗粒间的相互作用能(VTOT)由范德华引力作用能(VVDL)和双电层静电排斥作用能(VEDL)组成(公式2),VTOT的大小决定着胶体的稳定性,进而影响着胶体在水环境中的凝聚与分散行为[29]。其中,VVDL可通过公式3计算,VEDL可通过公式4和5计算[30]:
式中:a为纳米塑料的平均水力学半径(m);φ为悬液的Zeta电位(mV);ε0为真空介电常数(8.85×10-12 C2/J·m);ε为溶剂相对介电常数(水中:78.5);h为颗粒间的空间距离(m);C为德拜长度的倒数(m-1);A为Hamaker常数,1.0×10-20J[31];Na为阿伏伽德罗常数;T为绝对温度,298.15 K;e为元电荷电量,1.60 × 10-19C;I为离子强度;K为Boltzmann常数;Z为金属离子的价态;λ为介质的特征波长(100 nm)。
根据前人相关研究,在添加了溶解性有机质的混合胶体体系中,胶体颗粒之间除了范德华引力作用和静电排斥作用外,可能还存在其他重要的相互作用,如空间位阻作用等,影响胶体颗粒凝聚[4, 22-23]。相对于经典DLVO理论模型,扩展的DLVO(EDLVO)模型除了考虑胶体颗粒间的范德华引力作用和静电排斥作用外,也考虑了空间位阻效应对胶体凝聚的影响[23]。因此,本研究利用EDLVO模型进行IOM(20 mg/L)作用下PNs-IOM混合胶体聚集动力学过程的拟合。其中,VTOT的计算见公式6,VST(h)为颗粒间的空间相互作用能,通过公式7和8计算[30]:
式中:FST(h)为胶体颗粒间距离为h时的空间相互作用力(N);s为覆盖在PNs表面的有机物之间的距离(m);l为PNs表面有机质吸附层的厚度(m);s和l的取值参照Mao等[23]的相关研究。
1.1. 药品和试剂
1.2. 藻源溶解性有机质的提取与表征
1.3. 凝聚动力学实验
1.4. 仪器表征
1.5. 相互作用力理论计算
-
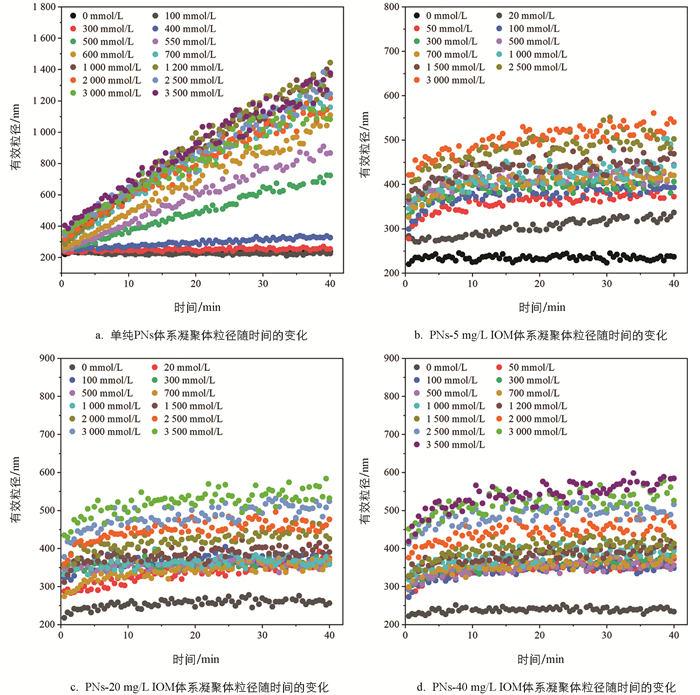
向PNs和添加不同质量浓度IOM的PNs-IOM胶体悬液中加入NaCl电解质,体系均会发生不同程度的凝聚,其凝聚体粒径随时间的变化如图 2所示。由图 2a可知,未添加IOM时,随着NaCl浓度的增加,PNs胶体凝聚体粒径增长速率逐渐加快。其中,当NaCl浓度小于300 mmol/L,凝聚体粒径随时间几乎不增长;随着NaCl浓度升高到400~700 mmol/L,凝聚体粒径随时间呈线性增长,且不同浓度间存在显著差异,例如400 mmol/L和700 mmol/L NaCl条件下,40 min后凝聚体粒径分别为327.5 nm和1 159 nm。这说明此浓度条件下PNs胶体双电层重叠所产生的静电排斥力较强,颗粒间有效碰撞概率小于1,对应慢速凝聚(即反应控制簇团凝聚,RLCA)阶段[32];随着NaCl浓度进一步升高,不同浓度条件下凝聚体粒径随时间的增长曲线趋于重合,说明此时PNs胶体颗粒间的双电层被强烈压缩,颗粒间的静电斥力显著减小,有效碰撞概率接近于1,进入快速凝聚(即扩散控制簇团凝聚,DLCA)阶段[33-35]。类似现象也在其他纳米塑料胶体颗粒(如聚对苯二甲酸乙二醇酯PET)聚集过程中呈现过[22]。
IOM的添加促使PNs的凝聚动力学过程发生明显改变,且伴随着体系阳离子浓度变化呈现不同的变化趋势(如图 2b-2d)。仅向PNs胶体悬液中添加0~40 mg/L的IOM并未显著改变胶体悬液的平均粒径(230 nm左右),说明PNs-IOM混合胶体也是稳定分散的;向混合胶体体系中加入低浓度NaCl(<300 mmol/L)溶液,与单纯PNs体系不同,混合胶体体系能迅速形成粒径为290~330 nm的凝聚体,且凝聚体粒径随时间缓慢增长,40 min后稳定在350~400 nm,说明IOM添加能促进低NaCl浓度条件下PNs胶体的快速凝聚,且IOM质量浓度越高,形成的凝聚体粒径越小;此后,随着NaCl浓度进一步升高,PNs-IOM混合胶体凝聚体粒径缓慢增加,对应RLCA凝聚阶段。然而,凝聚体粒径均显著低于相应NaCl浓度条件下单纯PNs胶体体系的凝聚体粒径,说明IOM的添加显著抑制了PNs胶体的凝聚,与前人研究HA对PNs凝聚的影响结果类似[34]。
-
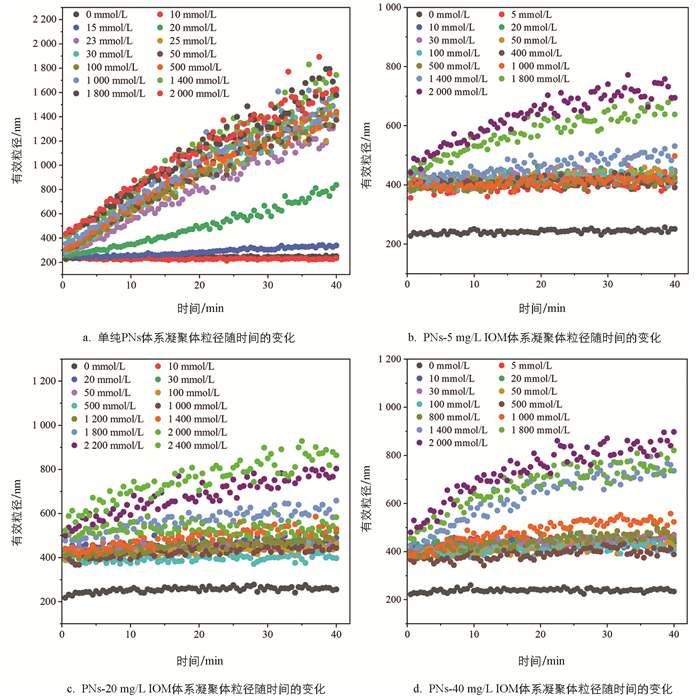
与NaCl体系类似,向PNs胶体悬液中加入不同浓度的CaCl2溶液,凝聚体粒径随时间呈现不同的变化趋势。由图 3a可知,当CaCl2溶液浓度低于15 mmol/L时,凝聚体粒径随时间几乎不增长;当CaCl2浓度为15~25 mmol/L时,PNs胶体凝聚体粒径随CaCl2浓度增加而增长,对应RLCA凝聚阶段;随着CaCl2浓度进一步升高,不同浓度间凝聚体粒径随时间的增长曲线趋于重合,进入DLCA凝聚阶段。
向PNs-IOM混合胶体中加入CaCl2溶液,混合胶体凝聚体粒径随CaCl2浓度的变化也呈现出与NaCl体系下相似的变化趋势(图 3b-3d)。向PNs-IOM混合胶体中加入较低浓度的CaCl2(<20 mmol/L)溶液能促使体系迅速形成粒径约为430 nm的凝聚体,此后凝聚体粒径随时间增长缓慢,40 min后粒径达到421~457 nm,显著高于相同CaCl2浓度条件下PNs胶体的凝聚体粒径(233 nm,图 3a),略高于低浓度NaCl条件下形成的凝聚体粒径(350~400 nm,图 2b-2d),说明IOM添加能促进低浓度CaCl2条件下PNs胶体的凝聚;此后,随着CaCl2浓度升高,PNs-IOM混合胶体凝聚体粒径缓慢增加,40 min后形成的凝聚体粒径(446~523 nm)显著低于相应CaCl2浓度条件下PNs胶体体系的凝聚体粒径(822~1 613 nm,图 3a),略高于相同浓度NaCl条件下PNs-IOM混合胶体的凝聚体粒径(图 2b-2d),说明添加IOM也能显著抑制高浓度CaCl2引起的PNs胶体凝聚,与Xu等[36]的研究结果类似。此外,CaCl2作用下,IOM添加量越高,PNs-IOM混合胶体形成的凝聚体最终粒径也越大,例如1 400 mmol/L CaCl2条件下,添加40 mg/L IOM后体系最终形成的凝聚体粒径(735 nm)显著大于添加5 mg/L IOM的体系(531 nm),这可能与Ca2+在PNs-IOM混合胶体颗粒间高效的架桥能力有关[22, 37]。
-
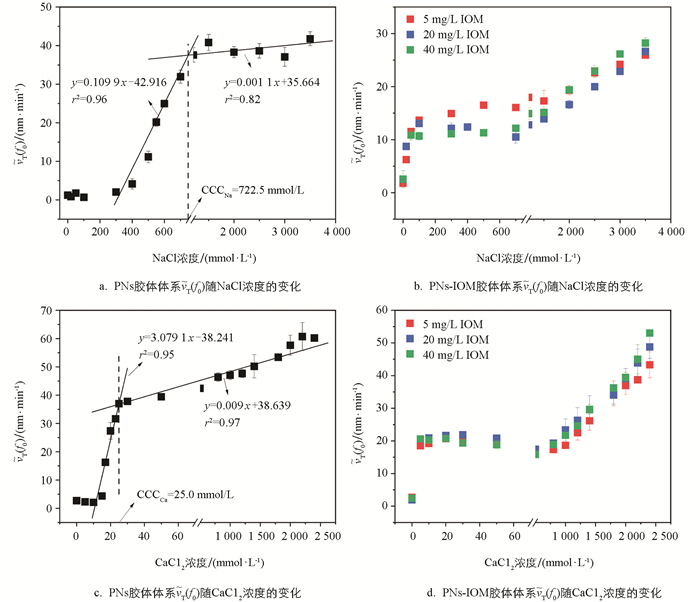
为了定量表征IOM对PNs胶体悬液凝聚能力的影响规律,本研究进一步计算了NaCl和CaCl2作用下胶体凝聚的
$\widetilde{v}_{T}\left(f_{0}\right)$ 。如图 4所示,未添加IOM时,随着NaCl和CaCl2浓度的升高,PNs凝聚的$\widetilde{v}_{T}\left(f_{0}\right)$ 值先直线上升,而后趋于平缓。这两种变化趋势分别对应着前文提到的RLCA和DLCA凝聚阶段,二者的交点所对应的电解质浓度即为临界聚沉浓度(Critical Coagulation Concentration,CCC)[23, 33]。因此,NaCl和CaCl2作用下PNs胶体凝聚的CCC值分别为722.5 mmol/L(图 4a)和25.0 mmol/L(图 4c),前者是后者的28.9倍。这说明二价阳离子Ca2+较一价Na+具有更强的屏蔽PNs颗粒表面负电场的能力,更加容易引起PNs胶体的凝聚[37]。向PNs胶体中加入IOM,IOM对PNs-IOM胶体凝聚的影响受到体系电解质浓度、类型和IOM质量浓度的共同调控。首先,电解质浓度会显著影响IOM添加后PNs-IOM的凝聚特征。IOM能显著促进低浓度电解质(NaCl和CaCl2)作用下PNs-IOM胶体凝聚,而抑制高浓度电解质作用下PNs-IOM胶体凝聚。当NaCl浓度<300 mmol/L或CaCl2浓度<20 mmol/L时,PNs-IOM胶体凝聚的
$\widetilde{v}_{T}\left(f_{0}\right)$ 分别稳定于约12 nm/min(图 4b)和20 nm/min(图 4d),显著高于未加入IOM的PNs胶体体系[NaCl:2 nm/min(图 4a);CaCl2:16 nm/min(图 4c)]。此后,尽管PNs-IOM胶体凝聚的$\widetilde{v}_{T}\left(f_{0}\right)$ 随NaCl和CaCl2浓度缓慢升高,其数值均显著低于相应电解质浓度条件下PNs胶体凝聚的$\widetilde{v}_{T}\left(f_{0}\right)$ 值,例如,NaCl浓度为3 000 mmol/L条件下,PNs+5 mg/L IOM胶体体系中的$\widetilde{v}_{T}\left(f_{0}\right)$ 值为24.2 nm/min(图 4b),远低于PNs体系中的37.1 nm/min(图 4a)。尽管已有大量文献研究了溶解性有机质HA或者FA对纳米塑料凝聚的影响[31-32],但是目前鲜见类似的电解质浓度效应报道。其次,电解质类型也会显著影响PNs-IOM胶体的凝聚。CaCl2作用下PNs-IOM胶体凝聚的$\widetilde{v}_{T}\left(f_{0}\right)$ 值远高于相同浓度NaCl作用下的$\widetilde{v}_{T}\left(f_{0}\right)$ 值,例如,电解质浓度20 mmol/L条件下CaCl2体系的$\widetilde{v}_{T}\left(f_{0}\right)$ 为21.6 nm/min(图 4d),NaCl体系则为8.7 nm/min(图 4b),说明二价阳离子Ca2+较一价Na+更容易引起PNs-IOM胶体的凝聚。这与Xu等[37]研究结果类似,与Na+相比,Ca2+通过阳离子架桥作用更能促进EPS胶体的团聚。最后,IOM质量浓度也是调控PNs-IOM胶体凝聚的重要影响因子。NaCl作用下(<1 000 mmol/L),添加量5 mg/L IOM时PNs-IOM胶体凝聚的$\widetilde{v}_{T}\left(f_{0}\right)$ 值(16 nm/min)显著高于20、40 mg/L(12 nm/min)(图 4b),说明IOM质量浓度越高,对PNs-IOM胶体凝聚的抑制作用越强。然而,对于CaCl2体系,高质量浓度IOM会明显促进PNs-IOM胶体凝聚,40 mg/L IOM作用下PNs-IOM胶体凝聚的$\widetilde{v}_{T}\left(f_{0}\right)$ 值(53 nm/min)显著高于20、5 mg/L IOM(43 nm/min)(图 4d)。 -
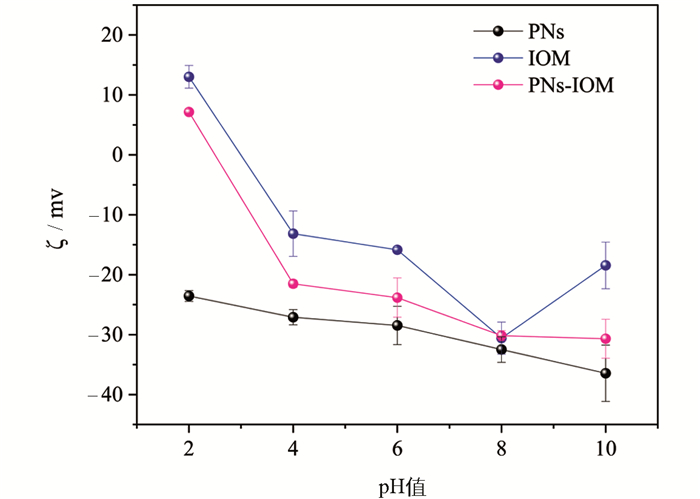
根据DLVO理论,胶体颗粒间存在静电排斥力和范德华引力,二者的相对大小决定着胶体的稳定性,进而影响着胶体颗粒在水环境中的迁移[30, 33]。其中,范德华引力仅与胶体颗粒自身性质有关,静电排斥力取决于胶体颗粒所带电荷密度,受到环境条件变化(如电解质浓度和类型、水体有机物等)的显著影响[29]。这意味着IOM和NaCl/CaCl2可能通过影响静电排斥力来影响PNs胶体颗粒的凝聚。图 5为不同pH值条件下PNs、IOM和PNs-IOM胶体的ζ电位。结果表明,在pH值为2~10的PNs胶体颗粒表面ζ电位均为负值,说明PNs表面带有大量净负电荷,体系等电点小于2.0。IOM表面整体也带有大量负电荷(pH值4~10范围内ζ电位小于0),IOM等有机组分较柔软且带有大量疏水基团,它们可以自发地通过一定疏水作用吸附并覆盖在PNs胶体颗粒表面[23],形成PNs-IOM混合胶体体系。该混合胶体体系的等电点约为2.5,在pH值为6时仍带有大量负电荷(ζ电位约为-23.8 mV),颗粒间静电排斥力较强,这也解释了本研究中仅添加不同质量浓度IOM并未引起PNs-IOM胶体凝聚的实验现象(图 2-4)。值得注意的是,在pH值为2~10时,PNs-IOM胶体体系的ζ电位绝对值均显著低于PNs体系,表明IOM的添加能显著降低PNs表面的负电荷。这意味着在相同的电解质浓度条件下,PNs-IOM胶体颗粒间的静电排斥力更小,理论上比PNs胶体更易凝聚。这一推论可以很好地解释低浓度NaCl(<300 mmol/L)或CaCl2(<20 mmol/L)条件下PNs-IOM胶体凝聚体粒径和凝聚速度均高于PNs胶体的实验现象(图 2-图 4),然而,却与高电解质浓度条件下PNs-IOM和PNs胶体凝聚特征不符。因此,可以推测在IOM存在下可能还存在其他作用力影响PNs-IOM胶体颗粒的凝聚。
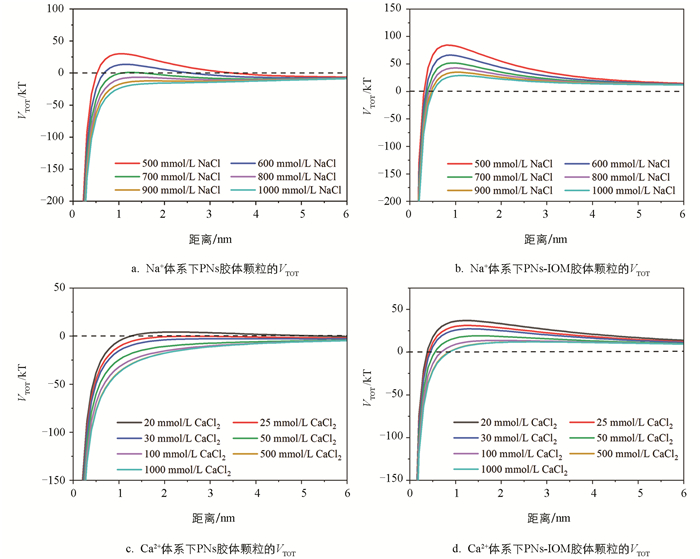
以往的研究表明,溶解性有机质(如HA和EPS)吸附在纳米塑料表面,除影响体系的表面电荷外,当有机质吸附层厚度超过颗粒间的德拜长度时,还可能通过产生强烈的空间位阻效应显著抑制纳米塑料胶体的凝聚[24, 31, 34]。在本实验中,IOM和PNs在pH值为6的条件下均带有负电荷(图 5),随着Ca2+和Na+浓度的进一步升高,除疏水作用外,PNs和IOM之间的静电排斥作用减弱明显,PNs表面吸附的IOM大分子组分可能更多,结构更紧密,进而产生更强的空间位阻效应,抑制PNs-IOM胶体颗粒间进一步地有效碰撞和凝聚。这也解释了本研究中IOM添加显著抑制高浓度NaCl和CaCl2作用下PNs-IOM胶体凝聚的实验现象。为了验证空间位阻及其对PNs-IOM胶体凝聚的影响,本研究进一步分别利用DLVO和EDLVO理论模型拟合分析了PNs和PNs-IOM胶体的相互作用能(图 6)。结果表明,在PNs胶体体系中,PNs颗粒间的能垒随着溶液中电解质浓度的升高逐渐降低,当NaCl浓度超过700 mmol/L或CaCl2浓度超过20 mmol/L时能垒基本消失(图 6a和图 6c),颗粒容易发生有效碰撞和快速凝聚,这与凝聚实验得到的CCCNa(722.5 mmol/L,图 4a)和CCCCa(25.0 mmol/L,图 4c)一致。证明在不添加IOM的体系中,PNs在NaCl和CaCl2溶液中的凝聚过程遵循DLVO理论,Na+和Ca2+主要通过压缩PNs胶体颗粒双电层,影响颗粒间的静电排斥作用力来调控PNs的凝聚与分散行为[38-39]。向PNs胶体中添加IOM,尽管PNs-IOM胶体颗粒间的能垒也随电解质浓度升高而逐渐降低,但其数值在浓度20~1 000 mmol/L范围内均大于0(图 6b和图 6d),且显著高于相应电解质体系下PNs胶体颗粒间的能垒(图 6a和图 6c)[31]。例如,当NaCl浓度为700 mmol/L时,PNs-IOM和PNs颗粒间的能垒分别为52 kT(图 6b)和1 kT(图 6a),前者是后者的52倍。更高的能垒意味着颗粒之间需要更大的动能才能克服斥力发生聚集[31]。该结果与高浓度Ca2+和Na+作用下PNs-IOM胶体难以发生快速凝聚,难以得到CCC值相一致(图 4b和图 4d)。说明在添加IOM的PNs-IOM体系中,胶体颗粒间的凝聚过程遵循EDLVO理论,除受静电排斥作用力的影响外,主要受颗粒间空间位阻效应的控制[23-24]。
对于不同的电解质,相对于一价Na+,二价Ca2+压缩PNs和PNs-IOM胶体颗粒表面双电层,降低颗粒间能垒的能力更强。例如,相同浓度(500 mmol/L)条件下,CaCl2体系中PNs胶体颗粒间的能垒(12 kT,图 6d)远远低于NaCl体系(84 kT,图 6b)。因此,Ca2+促进胶体凝聚的能力更强(图 2和图 3),凝聚速率更快(图 4)。此外,根据前人相关研究,在溶解性有机质添加条件下,Ca2+还可能通过架桥作用与吸附在纳米塑料表面的有机质发生反应,从而促进混合胶体的凝聚 [22, 37]。在本研究中,尽管IOM添加整体抑制了高浓度CaCl2作用下PNs-IOM胶体的凝聚,40 mg/L IOM作用下PNs-IOM胶体凝聚的
$\widetilde{v}_{T}\left(f_{0}\right)$ 值仍显著高于20、5 mg/L(图 4),这可能与高质量浓度IOM作用下,Ca2+的架桥作用位点更多,促进胶体凝聚现象更显著有关。由于Na+不具有架桥效应,IOM质量浓度越高,吸附在PNs表面越厚,产生的空间位阻效应越强,PNs-IOM胶体的凝聚速率越慢(图 4)。 -
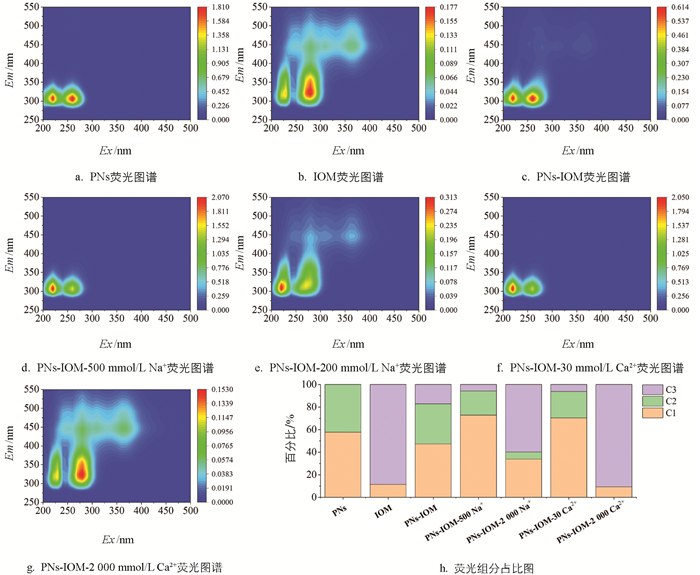
如前文所述,IOM通过疏水或静电作用吸附在PNs表面,可通过产生空间位阻效应影响PNs-IOM胶体的凝聚。考虑到IOM主要由蛋白质、氨基酸、多糖等大分子组成[21],本研究进一步利用三维荧光耦合平行因子法(EEM-PARAFAC)剖析了PNs-IOM胶体凝聚过程中的IOM各有机组分与PNs的相互作用(图 7)。通过分析所有样品的荧光图谱(图 7a-7g),得到3种主要的荧光组分(C1、C2、C3)及其占比,如表 1和图 7h所示。其中,C1(Ex/Em=220/320)为类色氨酸化合物[40]、C2(Ex/Em=260/320)为类色氨酸或酪氨酸化合物[40]、C3(Ex/Em=280/320)为微生物代谢副产物[41]。结果表明,在单纯PNs体系中,溶解性有机组分主要为PNs纳米颗粒和少量用于PNs分散的表面活性剂,集中在C1和C2部分(图 7a)。IOM的组分为C1和C3组分,其中C3组分占比达到89%(图 7b)。C3组分来源于大分子蛋白类有机物和小分子的酚类物质,主要是藻细胞的代谢产物和分解产物[41]。与PNs体系相比,PNs-IOM混合体系中C3组分由0%增加至17%(图 7h),C3组分分子量大且柔软,说明加入IOM后C3组分可迅速吸附在PNs胶体表面。在此基础上,向PNs-IOM混合体系中加入低浓度500 mmol/L NaCl和30 mmol/L CaCl2溶液,溶解性有机质中C2和C3组分的相对占比均显著降低(图 7h),说明低电解质浓度能促进PNs来源的C2和IOM来源的C3组分将优先通过架桥网捕或屏蔽颗粒表面负电场等作用吸附在PNs表面[42],促进PNs-IOM胶体凝聚形成如上文所述的290~430 nm疏松凝聚体(图 3和图 4)。向PNs-IOM混合体系中加入高浓度2 000 mmol/L NaCl和CaCl2后,C2组分和C1组分的相对占比减少甚至消失,说明此时更多塑料源的C2组分也吸附到了PNs胶体表面,通过形成较强的空间位阻抑制了胶体颗粒的进一步凝聚[37]。
-
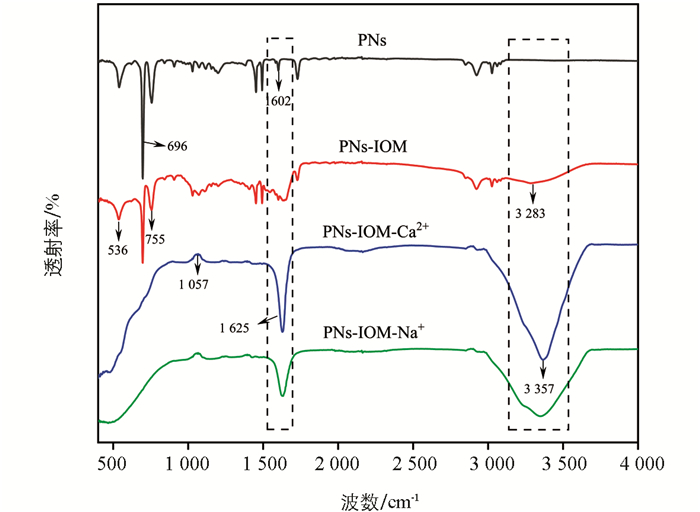
利用傅里叶变换红外光谱表征了胶体凝聚前后表面官能团结构的变化,如图 8所示。与单纯PNs体系相比,PNs-IOM体系特征峰峰值均减弱,如755、696、536 cm-1等处,表明IOM中C3组分吸附在PNs表面(图 7h),使得其芳香环结构发生了变化或含量有所减少[43]。此外,PNs-IOM体系在3 283 cm-1处出现新的吸收峰,归因于羟基(-OH)的伸缩振动,这表明IOM通过其表面的羟基或芳香环结构与PNs胶体进行相互作用[22],从而快速吸附于PNs表面。此外,与PNs-IOM体系相比,加入不同电解质后1 625 cm-1和3 357 cm-1处均出现了新的吸收峰,归因于芳香族C=C的伸缩振动和O-H的伸缩振动[23, 34],说明金属阳离子促进了IOM与PNs的相互作用,使得混合胶体表面的芳香物质增多并出现了新的酚类物质组分(图 7h);此外,1 057 cm-1处吸收峰强度增强,说明在PNs-IOM体系中加入Na+和Ca2+会促进更多的C-O键生成[37],这可能与塑料源或IOM源组分吸附在PNs表面有关。
2.1. 胶体颗粒凝聚体粒径随时间的动态变化规律
2.1.1. NaCl体系
2.1.2. CaCl2体系
2.2. 胶体颗粒凝聚速率和临界聚沉浓度的变化规律
2.3. 藻源有机质作用下胶体颗粒凝聚机理解析
2.3.1. 胶体颗粒间相互作用力的变化
2.3.2. IOM组分与PNs的相互作用
2.3.3. 胶体凝聚过程中体系官能团结构变化
-
1) Na+和Ca2+均可引起水溶液中PNs胶体的凝聚,阳离子浓度和价态越高,双电层压缩越严重,胶体颗粒间的静电排斥力越小,胶体凝聚所需克服的能垒越低,越易引起凝聚。添加Na+和Ca2+后PNs胶体颗粒凝聚先后经历RLCA慢速凝聚和DLCA快速凝聚阶段,且CCCNa(722.5 mmol/L)是CCCCa(25.0 mmol/L)的28.9倍。
2) 向PNs胶体中加入IOM,PNs-IOM胶体凝聚受到体系电解质浓度、类型和IOM质量浓度的共同影响,由静电排斥力和空间位阻效应共同调控。其中,PNs-IOM体系中低浓度Na+(<500 mmol/L)和Ca2+(<30 mmol/L)的添加会迅速促进IOM来源的C3组分和PNs来源的C2组分通过与PNs表面羟基或芳香环结构发生相互作用而吸附在PNs表面,通过降低胶体颗粒间的静电排斥势能促进PNs-IOM胶体凝聚,形成290~430 nm疏松凝聚体。然而,高浓度Na+和Ca2+的添加会使得更多PNs来源的C2组分快速吸附在PNs表面,通过形成强烈的空间位阻效应显著抑制PNs-IOM胶体颗粒间的进一步凝聚。与一价Na+相比,二价Ca2+能够通过架桥作用促进吸附在纳米塑料表面的有机质相互靠近,促进混合胶体的凝聚。