



 下载:
下载:
-
微孢子虫是一类专性细胞内寄生的单细胞真核生物,宿主范围广泛,能够寄生在几乎所有的无脊椎动物和脊椎动物上[1].微孢子虫的细胞核含有多条染色体,核糖体为原核型的70S核糖体.微孢子虫的基因组高度减缩,绝大部分编码基因丢失了内含子,基因及基因间区的长度均变短,如感染哺乳动物的兔脑炎微孢子虫的基因平均长度为1 080 bp,基因间区长度仅为129 bp[2].微孢子虫基因组的特殊性导致其编码基因的准确预测是一个亟待解决的问题.绝大多数真核基因具有多个PolyA位点,在形成成熟的mRNA过程中,外界环境的细微改变导致在mRNA的不同剪切位点进行选择性剪切和多聚腺苷酸化,这个现象叫做可选择性多聚腺苷酸化(APA). APA能够影响胞外信号刺激、生长与发育、细胞增殖和多种疾病的发生发展[3-4].多聚腺苷酸化是真核细胞内mRNA转录后处理形成成熟mRNA的一个重要步骤(mRNA转录处理的3个主要步骤分别是:5'帽子结构的形成,内含子的剪切和3'端加尾巴[5]),它影响着基因的表达,对预测基因结构有着巨大的作用.多聚腺苷酸化作用机制:切割及多聚腺苷酸化特异因子(Cleavage Polyadenylation Specific Factor,CPSF)绑定到PolyA信号序列,切割活化因子(Cleavage stimulation Factor,CstF)识别下游的U-rich和G/U-rich序列并相互作用,切割因子CFI在PolyA信号和下游作用元件之间的某个位置对前体mRNA进行分裂,最后在PolyA聚合酶的作用下添加多聚腺苷酸尾巴[6]. PolyA位点结构特征如图 1所示.
由于PolyA位点的预测对基因结构的分析和mRNA的形成机制有着重要的作用,近年来PolyA信号的预测引起了越来越多的关注.起初,人们基于线性判别函数的原理设计出了POLYAH[7];1999年,Graber等利用马尔科夫模型来预测PolyA位点[8];2003年,通过Erpin统计PolyA位点上下游的序列中一些位置特异的二核苷酸对所出现的频率进行预测[9];随后开始采用机器学习的方法来预测PolyA位点,例如基于SVM的PROBE[10];现在人们最常用的是POLYAR方法,它把PolyA位点分成3类(PAS-strong,PAS-weak,PAS-less)进行预测[11],还有研究者利用神经网络进行预测[12].然而这些方法却很少在微孢子虫基因组上尝试,主要原因在于人类基因与植物基因的研究颇为成熟,许多软件是针对研究较为成熟的人类基因或者植物基因表达偏好而设计的.目前已报道的微孢子虫有1 400多种,不同种属微孢子虫的基因组大小差异较大(2.3 Mbp~24 Mbp)[13-15].人类基因和植物基因相较于微孢子虫基因更为复杂,然而许多方法即使能够用于病原体的研究,结果也会存在较大误差.
本文以Encephalitozoon cuniculi的基因组为数据材料[16-17],对其进行分析,设计了微孢子虫PolyA信号的特异性预测算法.文中提出了一种新的特征提取方法,然后基于SVM机器学习算法来对PolyA信号进行预测.具体过程是通过运用一些特征表达方法,例如位置权重矩阵(PWM)[18]、k阶核苷酸出现频率来进行特征提取,运用主成分分析(PCA)法进行冗余特征的筛选,最后使用SVM分类器进行分类训练,从而建立微孢子虫PolyA信号的预测算法.
全文HTML
-
本研究从NCBI数据库中下载Encephalitozoon cuniculi的基因组序列作为数据集.数据分为训练集和测试集.扫描全部基因序列,得到348条含有AATAAA的片段,训练集和测试集的正集各取174条序列,余下的序列分别作为训练集和测试集的负集,经过实验取证,本文用174条序列作为训练集的负集,821条序列作为测试集的负集.并且对数据集中的每一条序列都进行处理,剪切形成以PolyA信号AATAAA为中心,前后各100 bp的核苷酸,共206个核苷酸长的序列.
-
现在比较流行的生物数据特征分析是机器学习领域的支持向量机(SVM)[19],SVM经常用于数据分类和回归问题.它的原理是将所有待分类的点映射到高维空间,然后在高维空间中找到一个能将这些点分开的“超平面”. SVM提供了一种避开高维空间的复杂性,直接用此空间的内积函数(核函数),再利用线性可分情况下的求解方法直接求解对应的高维空间的决策问题.当核函数已知时,可以简化高维空间问题的求解难度.支持向量机具有很好的泛化推广能力,它在生物信息领域的应用越来越广. SVM的核函数有3种:线性核函数、多项式核函数、高斯核函数[20].考虑到SVM具有良好的分类效果和在生物学中的广泛应用,文中将通过特征提取和空间降维所得到的数据,运用多项式核函数来进行分类.
-
PolyA的序列与其他序列(例如启动子)保守性较弱,使得很难分析PolyA位点的位置保守性.近几年位置权重矩阵(PWM)已经被广泛应用到分子生物学中,用来描述PolyA位点附近的碱基保守水平.构建位置权重矩阵时,横坐标代表 4个核苷酸碱基(A、T、C、G),纵坐标代表在这条基序中对应的位置信息,矩阵中的值代表每一个可能出现的核苷酸在对应位置的频率.计算公式如下:
ni,b(i=1,2,…,N;b=A,T,C,G)代表的是序列的第b个碱基在第i个位置上出现的次数,其中N是所对应的序列的碱基总数.
PWM被定义为
θ0,b(b=A,T,C,G)是序列上碱基b出现的随机频率,为了避免分母为0的情形,令θ0,b=0.253. fi,b是碱基b在位置i出现的频率.在这个算法中,用3-mer频率作为位置权重矩阵的参数.
k阶是长度为k(k=1,2,3,…)的核苷酸的低聚物.例如A为1阶核苷酸类型,CG为2阶核苷酸类型,ACG为3阶核苷酸类型.借鉴蛋白质序列的特征组织方式来形成特征,本算法中的特征提取只考虑1~3阶的核苷酸出现的频率.对PolyA信号前后的200 bp序列综合考虑,共得到84个特征,在此基础上,本文又提取了T在上游和下游序列出现的频率和G在下游出现的频率共得到87个特征数据.
提取得到的原始特征空间的维度很大,变量太多会增加计算量和问题的复杂性,为了更加全面系统地分析问题,在这里我们采用PCA的方法来进行空间的降维. PCA的计算步骤如下:① 计算相关系数矩阵;② 计算特征值和特征向量;③ 计算主成分载荷;④ 得出综合信息并进行递减排序,从而降低原始空间的维度.
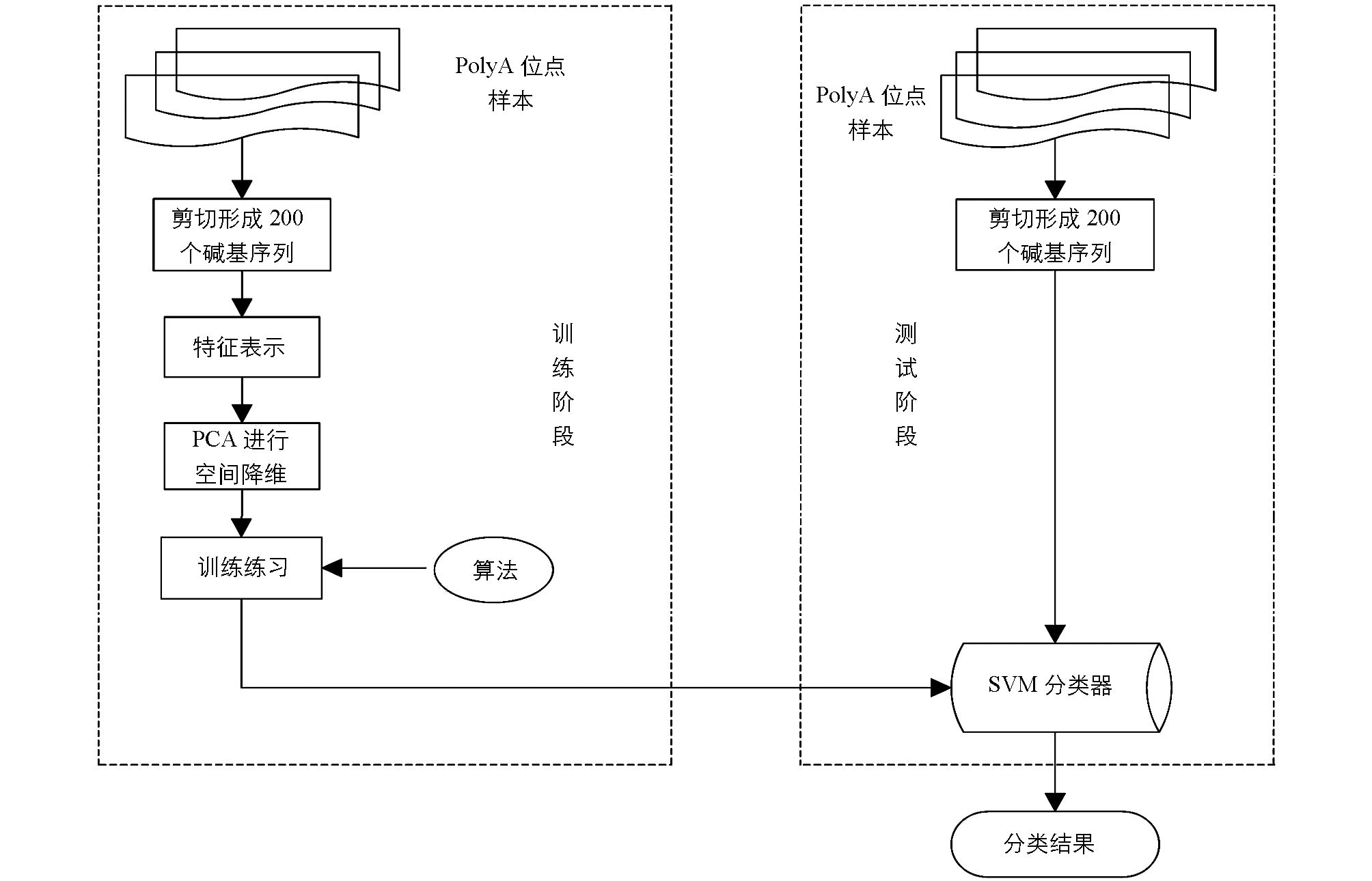
对于一个给定的微孢子虫PolyA位点,该PolyA位点预测基本流程图如图 2所示.
1.1. 数据集
1.2. 支持向量机(SVM)
1.3. 特征提取
-
本文通过敏感度(Sensitivity,Sn)、特异度(Specificity,Sp)[21]、准确度(Accuracy,ACC)、假阳性率(False Positive Rate,FPR)[22]和真阳性率(True Positive Rate,TPR)来评价模型的好坏.它们的定义如下:
其中:TP表示真阳性,FP表示假阳性,TN表示真阴性,FN表示假阴性.
ROC曲线的横纵坐标分别为FPR和TPR,它代表横纵坐标之间的协同变化关系,是一种分类模型的评判标准. ROC曲线被广泛应用在分类模型的评价指标中,它的精准率一般用曲线下的面积表示,曲线下的面积越小,表示模型越不精准.
-
经过特征提取得到原始空间的特征,然后用PCA进行降维,再用机器学习算法进行分类分析.分别对原始空间的特征和PCA降维后空间的特征进行分类,得出的特异度、敏感度、精确度大致相同.由于家蚕病原体的Encephalitozoon cuniculi的基因组序列的实验数据集的限制,本文使用原始空间的特征进行实验,采用SVM、决策树算法和KNN算法对本实验的数据进行分析比较,但是决策树算法和KNN算法分类出来的效果并没有SVM的好,SVM算法的精确度能达到85.14%,因此SVM算法有明显的算法优势. 表 1为微孢子虫PolyA位点识别算法的性能表.
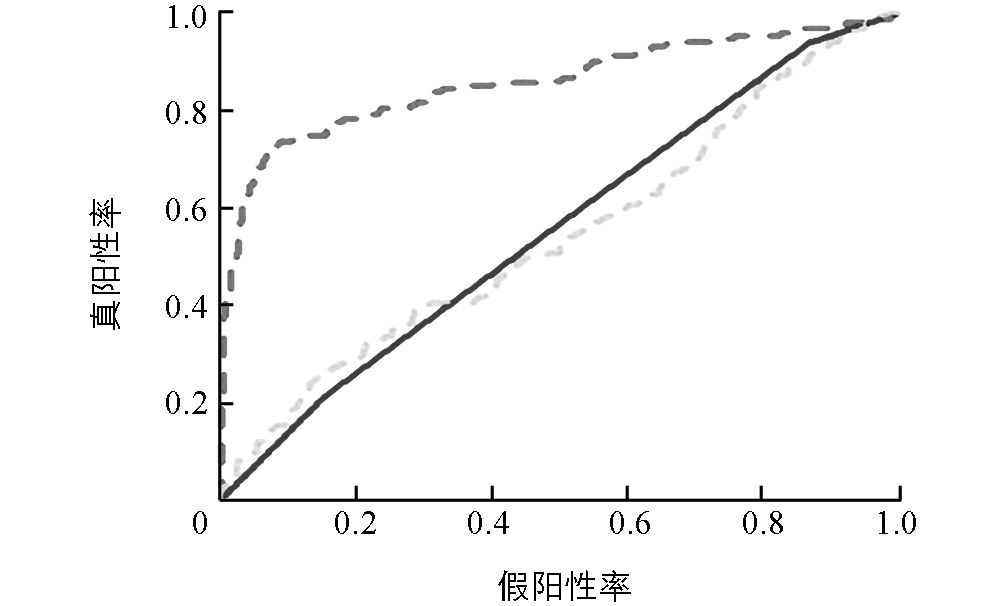
在相同数据的情况下,用ROC曲线来对SVM算法、决策树算法和KNN算法作分析,得到的结果如图 3所示,图中的红色虚线表示SVM算法,黄色虚线表示决策树算法,蓝色实线代表KNN算法.在微孢子虫PolyA位点的特异性预测算法中,SVM算法表现出了较高的性能.
2.1. 评价指标
2.2. 微孢子虫PolyA位点的识别算法比较
-
机器学习在选择性剪切位点预测、启动子的预测等应用中已经十分普遍,本文采用SVM机器学习的算法来对微孢子虫PolyA位点进行预测分析.为了使结果更加精确,根据PolyA位点附近的特征(下游是U,U/G丰富的区域),在提取1~3阶的核苷酸出现的频率的基础上又提取了T在上游和下游出现的频率以及G在下游出现的频率来作为特征数据.使用目前应用非常广泛的特征提取方法—位置权重矩阵(PWM),并运用核苷酸的三阶频率来计算位置权重矩阵.然后用PCA的方法进行数据降维,最后用机器学习中的SVM算法来进行分类,通过对模型的评估与实验,得出了一个较好的结果.家蚕病原体的Encephalitozoon cuniculi的基因组序列的数据集不像人类基因组序列的实验数据集那样的庞大、丰富,虽然通过PCA降维去除了一些冗余特征,但由于研究对象的实验数据的限制,本文采用PCA降维之前的特征进行实验,与决策树和KNN算法相比,SVM有较大的优势.由于微孢子虫的基因组并没有一个完整注释PolyA位点的数据库,虽然算法产生出一个良好的结果,但在这一点还是有限制的.
由于微孢子虫的基因库还不够完善,在这里用SVM算法对其进行分类预测,得出了较高的SnC,Sp和AC.相较于在水稻基因中的研究,本算法的精确度还是比较高的.在接下来的研究中,我们将更加深入地了解微孢子虫的基因结构特征,进而对算法进行改进,使之更加精确和实用.我们希望通过运用计算机知识对微孢子虫进行PolyA位点预测,为生物学微孢子虫的研究者提供一个更好的思路.