


 下载:
下载:
-
随着科技的进步,一些犯罪案件的科技含量也在提高,手段也在不断多样化,并且常规物证被有意识地销毁,往往给侦破工作带来难度,因此微量物证尤其是生物检材越来越引起物证工作者的重视. 2011年公安部关于《公安机关侦办命案工作规范》中要求,外围现场和关联现场要受到重视,尽可能多地发现并提取有用价值的物证,尤其是微量物证和生物检材.植物是犯罪现场中一种重要的环境和过程证据,而且很多案件中常常出现植物类物证,这些植物类物证大多都无法用形态学检验方法来鉴别,需要借助植物DNA技术手段对植物物证的来源和种类进行鉴定.这对于查找破案线索、划定侦查范围和方向提供了有价值的证据,也为法庭诉讼产生重要的指导和支撑作用[1-2].近年来,CBOL(生物条形码协会)植物工程组对约550个物种进行通用引物筛选,试图用一个或组合的DNA条形码来鉴别植物物种,这些研究成果已在鉴别违禁植物、毒品、出入境物种资源等方面得到应用[3].植物物证个体识别方面的研究报道不多,虽然也有用传统DNA分子标记技术进行DNA多态性比对分析,进行同一认定的成功案列,但该方法难以实现自动化,检测方法相对缓慢而且费力.目前,利用RAD-seq技术来进行植物物证的个体识别还未见报道.
简化基因组测序是在第二代测序技术基础上发展起来的,是利用酶切技术、序列捕获芯片技术或其他实验手段降低物种基因组复杂程度,针对基因组特定区域进行测序,进而反映部分基因组序列结构信息的综合实验技术[4],目前运用较广泛的方法是限制性酶切相关位点的DNA测序(Restriction Association site DNA sequencing,RAD-seq)[5]. RAD-seq所得的全基因组范围特异酶切位点附近的小片段DNA标签,能较好地代表整个基因组的序列特征,从而能够在大多数生物中获得成千上万的单核苷酸多态性(Single Nucleotide Polymorphism,SNP)标记. RAD-seq技术操作简单,不受参考基因组限制,并可简化复杂基因组,目前已广泛应用于分子育种、系统进化、种质资源等领域[6-8],但在植物物证个体识别方面,RAD-seq应用于法庭科学中具体的实验实施和分析方法还需要摸索细化.
桂花Osmanthus sp.在重庆地区分布很广,在公园、路边、居民区随处可见,桂花可以年年开花结果,且花期较长,与我们的日常生活紧密联系,如果涉案出现的可能性较大.本文的研究对象选择没有可参考基因组的桂花作为植物物证个体识别的样本,利用简化基因组测序技术来降低基因组测序和分析的复杂度,进而构建植物物证桂花样品的个体识别数据库,试图找到能识别植物物证样品桂花同一个体的方法,为后续利用RAD-seq技术进行植物类物证的个体鉴别提供可靠的方法和支持.
全文HTML
-
植物物证桂花样品全部于2015年10月采自西南大学校园内,共随机采集15棵桂花树上的叶片,每个样品随机采集2~4片叶片,分别装入自封袋中,按1~15顺序号标注,置于4 ℃冰箱备用.,
-
提取叶片样品基因组总DNA进行分析.把随机采集的15棵桂花树叶片,按序号随机从中取叶片0.5克,在CTAB(Hexadecyltrimethy Ammonium Bromide)法[9]的基础上略做改进以提取植物总DNA.采用NanoDroP 2000测定A260/A280比值评价其DNA浓度及纯度.
-
选择EcoR1六碱基限制性内切酶对基因组DNA样品进行酶切,保证产生的RAD标记能够在基因组上均有分布,同时获得的RAD标记数量能够达到实验所需的饱和度.将PCR后产物进行DNA片段回收,并对最终构建完成的文库进行检测.测序仪器为IlluminaHiseq4000.对所测得的原始数据进行过滤,过滤标准为:所有SNP位点总深度须大于等于4,如果SNP为杂合型则次好碱基深度须大于等于2.过滤后的序列根据index序列划分到具体个体,便于后续分析.
-
二代高通量测序获得15个已知桂花样本及3个未知植物物证样品桂花的原始DNA序列,本文依据如下标准对原始数据进行过滤:① 仅使用含有Hind Ⅲ酶切识别位点Read1序列;② 利用Q30标准对序列质量进行评估;③ 所得的序列的前50 bp不存在不确定碱基;④ 整条序列中不确定碱基不多于3个,过滤后reads数目比例如图 1所示.从数据量的整体上看,数据有效率高达97.28%,经过滤后共获得6,638 bp,587,602 bp的Clean data,数据量统计如表 1所示.
-
利用RAD技术对过滤后的数据进行聚类和SNP查找,并对15个样品通过聚类获得的初始SNP进行初步过滤,得到SNP数量为1197085个(表 2).
McCarroll等[10]认为群体遗传上的差异主要是通过对其群体上SNP位点信息的分析,来进行群体间遗传多样性的分析.植物物证桂花样品SNP数量从3373到158 467不等,样品平均SNP数量为79 805个. SNP数量最多的是植物物证桂花样品9,最少的是植物物证桂花样品8.每个个体的SNP差异性都反映了个体的多态性.杂合度最高个体是植物物证桂花样品4,高达83.52%.
-
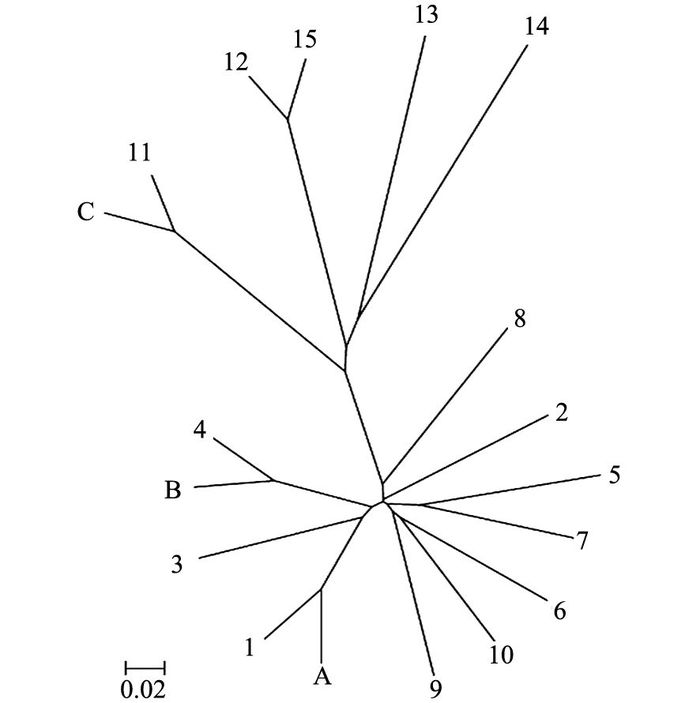
通过RAD技术获得18个(15个植物物证已知样品和3个未知植物物证样品)桂花SNP数据,然后对SNP进行筛选(筛选原则:纯合SNP深度至少为4,杂合SNP次好深度至少为2),最后依据至少在18个植物物证样品都存在的SNP位点(118) 和至少在17个植物物证桂花样品都存在的SNP位点(797) 总信息,计算样品的遗传距离,利用MEGA6构建系统发育树(图 1).
从图 1中可以得出,未知的A植物物证样品与桂花1样品最为接近,未知的B植物物证样品与桂花4样品最为接近,未知的C植物物证样品与桂花11样品最为接近,从而可初步估计A,B,C 3个植物物证样品分别对应桂花1、桂花4和桂花11这3个样品.而样品12和样品15在很接近的同一分支上,笔者推测:① 它们存在很近的亲缘关系;② 所在分支取样稀疏、从而相对位置接近.
-
为进一步解析并验证系统发育树分析结果,本文进一步通过建立SNP数据库和未知植物物证样品的SNP位点比较来找出具有区分效力的SNP子集.通过RAD技术获得15个桂花样品的SNP数据集,并对此数据集进行筛选,获得1 182个SNP位点,然后用这些数据集建库(筛选原则:纯合SNP深度至少为4,杂合SNP次好深度至少为2),将至少在14个植物物证样本都存在的SNP位点挑选出来建立植物物证桂花样品的SNP数据库.
通过RAD技术分别获得3个未知样品的SNP数据集,和以上原则一样进行筛选,然后将得到的SNP位点信息与SNP库做比对,最终得出统计结果(表 3).
从表 3中可以看出,未知植物物证样品A的SNP位点信息与桂花数据库比对后,桂花1与未知植物物证样品A的比对符合度最高,有148个位点符合,符合率达到12.52%;未知植物物证样品B的SNP位点信息与桂花数据库比对后,桂花4与未知植物物证样品B的比对符合度最高,有116个位点符合,符合率达到9.81%;未知植物物证样品C的SNP位点信息与桂花数据库比对后,桂花11与未知植物物证样品C的比对符合度最高,有112个位点符合,符合率达到9.48%.从而推测出A,B,C这3个未知样品分别是桂花1、桂花4、桂花11,与实际情况吻合.
-
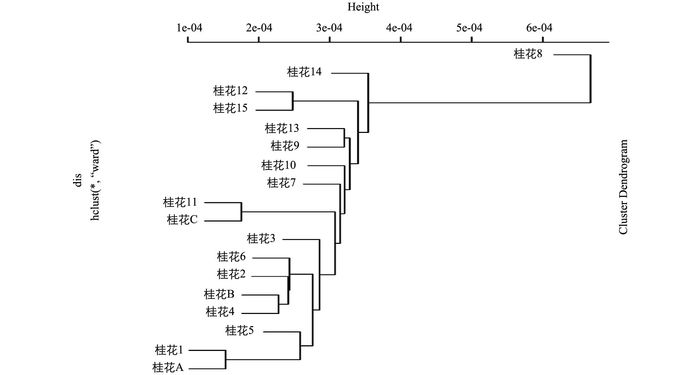
RAD-seq测序的片段是有RAD酶切后的产物,过滤后的clean data两端含有相同的酶切位点.先切除reads两端的酶切序列(reads1切除5 bp,reads2切除15 bp),从而避免酶切序列造成kmer偏好性.然后以9 bp(kmer)为滑动窗口,用jellyfish计算所有reads kmer出现的频数[11],考虑到样品测序量的不同,将频数转化成频率.根据所得的频率表用欧式距离公式计算样品间的差异度.距离越大,表示2个样品的遗传相似度越远.最后利用差异度矩阵通过离差平方和的方式将样品聚类,2个物种的差异度越小,在聚类图中的位置越近.欧式距离算法特性是高频kmer在计算样品差异度中起主导作用,但是某些序列的特性(如poloA)和PCR扩增过程等因素会造成某些kmer的频数急剧增大,干扰物种鉴别结果.因此,在计算差异度之前,我们过滤掉高频kmer(大于0.005%).
从图 2中可以看出,盲测的3个样品都能很好地和对照组聚类. A样品与样本桂花1聚类后距离最近,B样品与样本桂花4聚类后距离最近,C样品与样本桂花11聚类后距离最近.而且3组盲测样品聚类的顺序都先于其他样品,说明3组样品的遗传距离是最近的,聚类结果能很好地和实验设计吻合.
4.1. 测序基本信息分析
4.2. SNP的查找检测和建库
4.3. 系统进化树分析
4.4. 建库比对分析法
4.5. 用jellyfish计算法绘制聚类图分析
-
本文建库提取的植物桂花总DNA选取的材料是新鲜叶片,是一种理想状态,虽然本文首次尝试利用RAD-seq技术来识别植物物证桂花个体,并通过3种分析方法都可以成功识别未知的植物物证桂花个体,鉴定结论成立,存在同一性联系,但是在实际案件中植物材料不是干枯残缺就是量小且已破碎、腐败,肉眼只能鉴别是否植物检材的粗浅辨识,因此微量残败植物物证DNA提取方法的成熟建立也是满足下游实验的重要工作.本文作者也对植物基因组DNA提取方法进行了比较,发现改良的CTAB法比一般的植物DNA提取试剂盒获得总DNA的产量多,且纯度高,但是此方法操作步骤复杂,耗时长.在今后的工作中,要针对不同植物物证样本的状态和部位来建立合适的提取方法,满足实战需要.
二代测序技术在法庭科学领域中应用发展比较滞后,尚处于刚刚起步阶段[12-13].本文从3种分析方法都得到了相同的结果,即未知植物桂花样品A,B,C对应库中个体桂花1,4,11.在建库比对分析法的严格与宽松2种条件下,聚类图分析法显示3个植物未知样品都与桂花8的匹配度最低.这说明虽然分析方法的角度不同,但是数据内在的关联性被很好地反映了出来,进一步说明了数据分析的可靠性.相比较而言,基于k-mer频谱的方法采用了jellyfish软件、欧式距离等数学手段对数据进行处理和计算,减少了数据分析的误差,更易于推广到公安植物物证鉴别应用中.
从分析流程来看,还不能对个体鉴定是否存在阈值和是否有物种特异性做出评判.比如在建库比对分析中,未知植物样品A与桂花1有148条SNP位点信息相吻合,符合度达到了12.5%,这12.5%的吻合度是否可以对未知样品A与桂花1的同一认定做出定性的判断,在今后不同物种个体识别的研究中,是否可以找到一个最低阉值来做定性判断标准,基于k-mer频谱的方法给出了通过聚类距离远近来进行同一认定,但这个距离近到多少才认为是同一个个体也没有明确指出.这些都有待于下一步的研究,这项技术要被普及应用到法庭科学,还需要国际学者的广泛参与,建立公开明了的工作原理,优化完善技术流程,保证数据风险可控及价格稳定等,才可能被法庭和立法机构接受.
-
RAD-seq技术结合该文的分析方法能够识别出未知植物物证样品A,B和C分别对应于桂花1,4和11,实现对案件中关联植物物证进行精确检验鉴定的方法和一个小型鉴定实例,但其识别能力有待扩展到更大样本集和更多物种中.