


 下载:
下载:
-
植物叶面的微生物群落非常多样化,包括细菌、真菌、酵母等,这些微生物中的一些可能对植物有害,被称为病原体,而其他的可能因促进植物生长和发育对植物有益.微生物的活动是影响植物生长发育的关键因素[1-2].其中,细菌是叶面环境中最主要的微生物类群[3].研究发现,植物叶面细菌能产生激素促进植物生长、有助于减少化肥和农药投入、轻环境污染、对重金属产生耐受性和抗性,实现农业的可持续发展[4-7].例如:解淀粉芽孢杆菌在大田条件下对核盘菌引起的茎干枯萎病起到良好防治作用[8];苏云金芽孢杆菌也是植物叶际附生菌,可以保护植物免受草食昆虫的侵害[9];菌株D5/23T是一种很好的促植物生长菌,能产生植物生长激素和细胞激动素,并且能固定大气中的氮气,促进植物根和茎干生长,最终使不同的作物增加产量[10-12].
烟草作为经济作物和模式植物,叶面存在大量的微生物,这些菌群影响着烟草整个生育时期及后期的烘烤调制过程,对烟草品质的形成有着重要作用[13-15].而调制是烤烟生产中的重要环节.微生物在鲜烟叶调制过程中对烟叶产品的形成及其品质产生了不同的作用.其中,细菌作为烤烟调制中的主要微生物,研究者对其在烘烤过程中的作用进行了研究.研究发现,不同部位烟叶烘烤过程中的优势微生物种类不完全相同,优势细菌属11种,其中棒杆菌属(Corynebacterium)、肠杆菌属(Enterobacter)、芽孢杆菌属(Arthrobacter)、泛菌属(Pantoea)出现频率最高[16].烟叶在烘烤进程中,细菌种类一直在减少,从烘烤开始到结束,减少10~100倍[17-18].不同烘烤方式和烘烤条件下,细菌总量在烤烟烘烤开始后逐渐增多,到48 h时烟叶变黄前后达到最大,细菌种类随烘烤时间的增加而减少[19].某些微生物在烘烤过程中对烟叶的腐烂和烟叶有害成分含量的消长等方面有重要影响[16, 20-21].微生物在烟草烘烤阶段对烟叶中烟碱含量有一定的降解[22-24].到目前为止,大多数的研究都是关于发酵过程中的微生物,对于调制过程中的微生物,特别是细菌研究较少.对于调制过程中不同阶段细菌种属的多样性分析,能更好地促进有益细菌的利用,以用于烟草叶片的品质提高.然而前人分析微生物多样性的方法一般采用微生物平板纯培养方法、微平板分析方法、磷脂脂肪酸法以及DGGE等分子生物学方法,传统的方法只能分析可培养微生物,且存在误差,操作繁琐.
高通量测序技术对16S rDNA/18S rDNA/ITS等序列进行测序,能同时对样品中的优势物种、稀有物种及一些未知的物种进行检测,获得样品中的微生物群落组成以及相对丰度,在植物、土壤、肠道中广泛应用[25-28].本实验利用高通量测序技术对烘烤中烟叶细菌群落结构的多样性进行分析,旨在准确检测调制不同阶段细菌的种类.
全文HTML
-
AxyPrepDNA凝胶回收试剂盒(Axygen公司);QuantiFluorTM-ST蓝色荧光定量系统(Promega公司);TruSeqTM DNA Sample Prep Kit,OMEGA-soil DNA Kit(Omega Bio-Tek公司);agarose gels(biowest公司);FastPfu Polymerase(Trans Gen公司);TruSeqTM DNA Sample Prep Kit(Illumina公司).
-
PCR仪(ABI GeneAmp® 9700型);高速台式冷冻离心机(Eppendorf公司);电泳仪(北京市六一仪器厂);MISEQ测序仪(Illumina公司);HISEQ测序仪(Illumina公司);酶标仪(Biotek公司);微型荧光计(Turner Bio Systems公司);Covaris M220(Gene Company Limited公司);旋涡混合器(海门其林贝尔仪器制造有限公司);粉碎研磨仪(上海万柏生物科技有限公司).
-
在重庆市彭水烟叶基地单元种植云烟87.烟叶成熟采收后,选择质量较好、无病害的中部烟叶.选好烟叶后统一编杆,分别放置于不同的3个烤房(分别命名A,B,C烤房).然后分别在成熟采收(0 h)、变黄阶段的变黄前期(A,B,C烤房取样时间:16 h,18 h,17 h)、叶片变黄期(A,B,C烤房取样时间:33 h,41 h,29 h)、支脉变黄期(A,B,C烤房取样时间:52 h,66 h,41 h)、定色阶段的主脉变黄期(A,B,C烤房取样时间:77 h,77 h,65 h)、香气物质合成期转火节点(A,B,C烤房取样时间:105 h,94 h,116 h)取样.
-
根据E.Z.N.A.® soil试剂盒(Omega Bio-tek,Norcross,GA,U.S.)说明书进行总DNA抽提,DNA浓度和纯度利用NanoDrop2000进行检测,利用1%琼脂糖凝胶电泳检测DNA提取质量;用338F(5′-ACTCCTACGGGAGGCAGCAG-3′)和806R(5′-GGACTACHVGGGTWTCTAAT-3′)引物对V3-V4可变区进行PCR扩增,扩增程序:95 ℃预变性3 min,27个循环(95 ℃变性30 s,55 ℃退火30 s,72 ℃延伸30 s),最后72 ℃延伸10 min (PCR仪:ABI GeneAmp® 9700型).扩增体系为20 μL,4 μL 5×FastPfu缓冲液,2 μL 2.5 mmol/L dNTPs,0.8 μL引物(5 μmol/L),0.4 μL FastPfu聚合酶;10 ng DNA模板.
-
使用2%琼脂糖凝胶回收PCR产物,利用AxyPrep DNA Gel Extraction Kit (Axygen Biosciences,Union City,CA,USA)进行纯化,Tris-HCl洗脱,2%琼脂糖电泳检测.利用QuantiFluorTM-ST(Promega,USA)进行检测定量.根据Illumina MiSeq平台(Illumina,San Diego,USA)标准操作规程将纯化后的扩增片段构建PE 2×300的文库.
构建文库步骤:1)连接“Y”字形接头;2)使用磁珠筛选去除接头自连片段;3)利用PCR扩增进行文库模板的富集;4)氢氧化钠变性,产生单链DNA片段.利用Illumina公司的Miseq PE300平台进行测序.
-
原始测序序列使用Trimmomatic软件质控,使用FLASH软件进行拼接:
1) 设置50 bp的窗口,如果窗口内的平均质量值低于20,从窗口开始截去后端碱基,去除质控后长度低于50 bp的序列;
2) barcode需精确匹配,引物允许2个碱基的错配,去除模糊碱基;
3) 根据重叠碱基overlap将两端序列进行拼接,overlap需大于10 bp.去除无法拼接的序列.
使用的UPARSE软件(version 7.1 http://drive5.com/uparse/),根据97%的相似度对序列进行OUT(Operational Taxonomic Units)聚类;使用UCHIME软件剔除嵌合体.利用RDP classifier(http://rdp.cme.msu.edu/)对每条序列进行物种分类注释,比对Silva数据库(SSU123),设置比对阈值为70%.
1.1. 试剂和仪器
1.1.1. 试剂
1.1.2. 仪器
1.2. 方法
1.2.1. 烟叶样本采集
1.2.2. DNA抽提和PCR扩增
1.2.3. Illumina Miseq测序
1.2.4. 数据处理与分析
-
利用Miseq测序,分析了A,B,C 3个烤房,6个时间点(包含3个重复,烘烤前期为取样点1,2,3,烘烤后期为取样点4,5,6)共54个样本的细菌群落结构.首先,我们获得了3个烤房取样样本的测序条数(表 1).结果显示,从各个烤房的前、后期来看,后期OTU总量和丰富度指数有增高,多样性指数也增高.
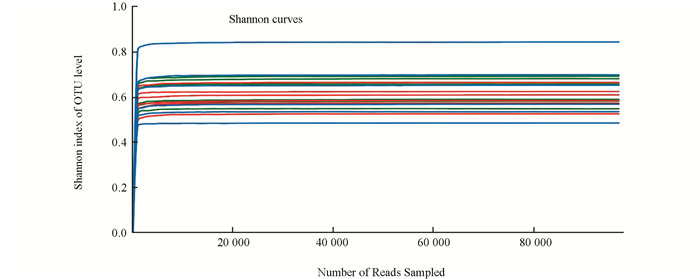
稀释曲线主要利用各样本的测序量在不同测序深度时的微生物多样性指数构建曲线,以此反映各样本在不同测序数量时的微生物多样性.选择97%相似度的OTU或其他分类学水平,利用mothur计算不同随机抽样下的多样性指数,利用R语言工具制作曲线图(图 1),从稀释曲线可以看出,按照测序数据量,已达到平台期,在此测序量下可以相对真实地反映菌群结构.
-
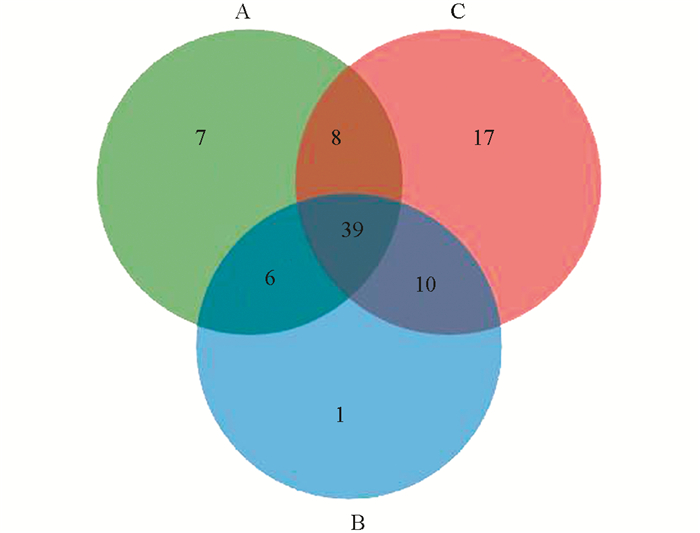
在OTU分析的基础上,选取相似水平为97%的OTU进一步进行了物种组成分析.通过利用Venn图展现A,B,C 3个烤房共有和独有的物种数目,直观展现不同烤房调制过程中烟叶的物种数目组成相似性及重叠情况.结果显示,A,B,C 3个烤房的OTU大部分都是共有的,其中B烤房独有OTU较少,C烤房独有OTU较多(图 2).
-
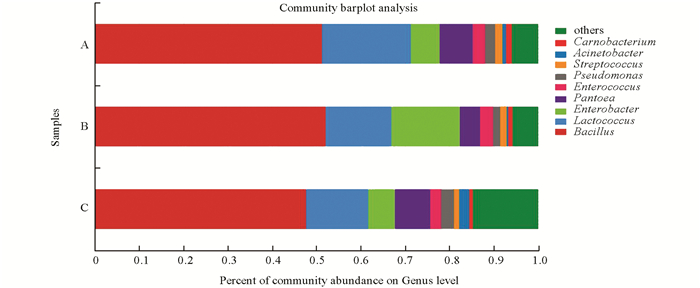
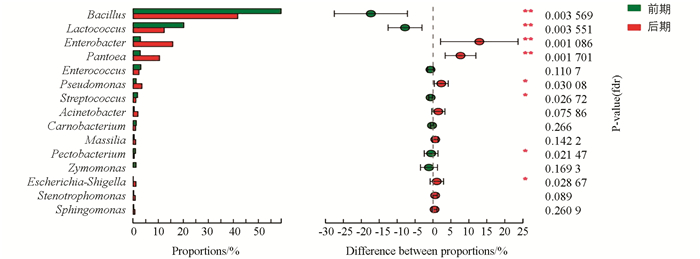
根据分类学分析结果,可以得知3个烤房在调制过程中各个烟叶样本在各分类水平上的群落结构组成情况.根据分析结果,可以获得各样本在某一分类学水平上含有何种细菌,以及烟叶样本中各细菌的相对丰度.结果显示,在3个烤房中,主要的属为芽孢杆菌属(Bacillus)(占比50%),其次为乳球菌属(Lactococcus)和肠杆菌属(Enterobacter).从图 3中看出优势的物种百分比相差不大,其中肠杆菌属在B烤房样本中比例比另外2个高(图 3).
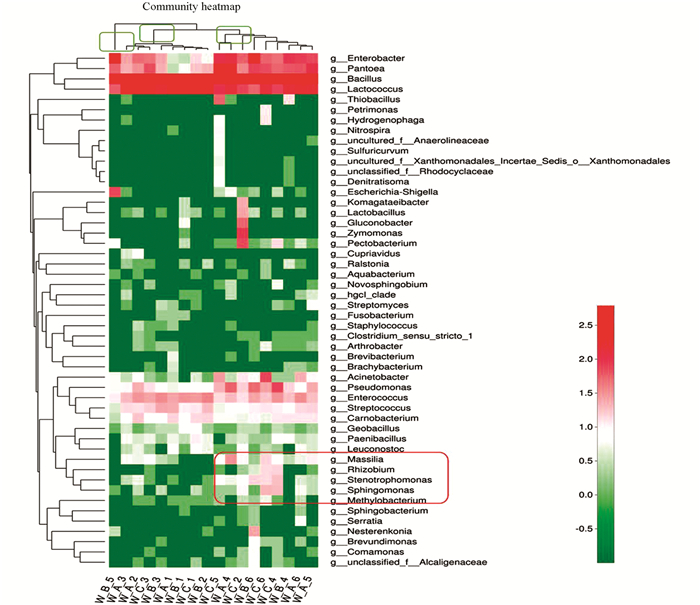
根据物种或样本间丰度的相似性进行聚类,并将结果呈现在群落heatmap图上(图 4),可使高丰度和低丰度的物种分块聚集,通过颜色变化与相似程度来反映不同样本在各分类水平上群落组成的相似性和差异性.通过分析发现,样本分成3类,如图 4上绿色框所示,一个框为一类;主要有两类,一类包含的是前期的3个烤房样本,一类是后期的3个烤房样本;前期、后期主要在红色框中物种差异较大,如马赛菌属(Massilia)、根瘤菌属(Rhizobium)、寡养单胞菌属(Stenotrophomonas)、鞘氨醇单胞菌属(Sphingomonas)等.
-
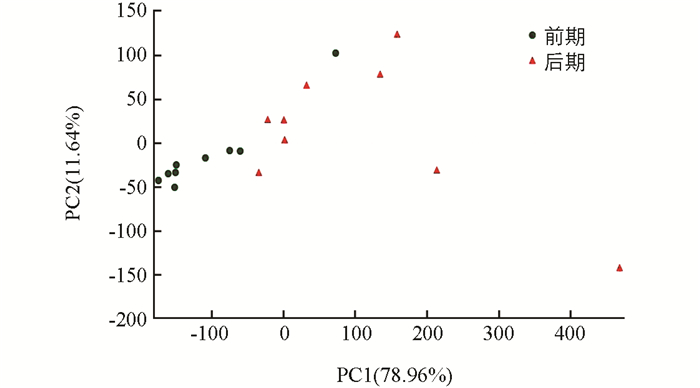
在物种组成分析的基础上,可以利用主成分分析(PCA分析)来比较确定影响细菌差异的因素,样本物种组成越相似,反映在PCA图中的距离越近.通过分组,将烘烤前期分为一组,烘烤后期分为一组.分析结果显示,烘烤前期样本聚集在一起,烘烤后期样本聚集在一起,有比较明显的区分,前期和后期细菌组成差异明显(图 5).
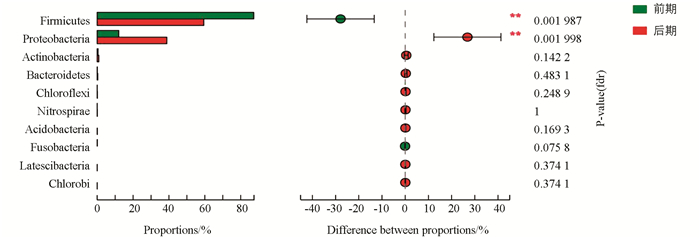
利用组间比较分析,根据PCA得出的结果,分析在主要影响因素下的差异物种.对前、后期两组进行wilcoxon秩和检验差异分析门和属水平,10个门中差异有统计学意义的为硬壁菌门(Firmicutes)和变形菌门(Proteobacteria),且后期Firmicutes下降,Proteobacteria上升.丰度前15的属中差异显著的为芽孢杆菌属(Bacillus)、乳球菌属(Lactococcus)、肠杆菌属(Entrobacter)、泛菌属(Pantoea)等(图 6,图 7).
2.1. 物种丰富度和多样性
2.2. Venn图比较
2.3. 细菌群落结构比较
2.4. 主成分分析
-
植物叶片上生存着大量不同性质的微生物,包括有益和有害微生物,它们与植物长期共同生存,组成了独特的微生物群落,形成植物微生物生态体系.许多研究证实,植物体内外的大量微生物与植物生长发育、植物抗性密切相关[4-6].前人关于微生物的鉴定分析多采用传统培养方法[29].近年来,高通量测序在微生物多样性分析上得到了广泛的应用[25-28].该技术相较于传统的纯培养方法及以16S rDNA为基础的非培养方法,能够产生覆盖深度更大的数据量,检测到纯培养和非纯培养技术未能发现的低丰度植物细菌种类,为丰富植物微生态学理论及基因工程菌的研究奠定基础[30].
烟草是我国重要的经济作物和模式植物,在烟叶生长过程中,环境中的微生物与烟草形成了植物-微生物共生体系[1].调制是烤烟生产中的重要环节,其中烟叶细菌的活动对烤烟调制过程和烟叶品质的形成有重要影响[19].为了快速、准确地分析烘烤过程中优势或致病细菌种群,本研究采用高通量测序,分析烘烤过程烟叶中细菌的群落结构.分析结果显示,在3个烤房中,芽孢杆菌属(Bacillus)占绝对优势.已有研究表明,芽孢杆菌是一种较为理想的生防微生物,易从土壤和植株中分离得到.目前,用于防治植物病害的芽孢杆菌主要有枯草芽孢杆菌(Bacillus subtilis)、侧孢芽孢杆菌(B.laterosporus)、蜡样芽孢杆菌(B.cereus)、地衣芽孢杆菌(B.licheniformis)、苏云金芽孢杆菌(B.thuringiensis)、多粘类芽孢杆菌(Paenibacilluspolymyxa)、短小芽孢杆菌(B.pumilus)[31, 32]等.进一步分析发现,烘烤前期和烘烤后期细菌组成差异明显,根瘤菌(Rhizobium)主要跟植物病害生物防治相关[33],而寡养单胞菌(Stenotrophomonas)、鞘氨醇单胞菌属(Sphingomonas)与植物的联系也非常密切,能降解有机农药和高分子有机污染物[34-35],鞘氨醇单胞菌属(Sphingomonas)还能在根际分泌糖类营养物供植物吸收[36],接种该菌可提高植株高度,增强果实干质量,进而促进植物生长[37].这些菌群的变化可能是由于烟叶在烘烤过程中,烘烤后期较前期内部物质发生了剧烈的变化而引起的.本研究利用高通量测序,全面分析了调制过程中烟叶细菌的群落结构,为进一步针对性地利用这些优势细菌以提高烘烤烟叶质量奠定了基础.