




-
竹亚科(Bambusoideae)为禾本科(Poaceae) 12个亚科中最大的亚科之一,因竹杆具有木纤维而不同于其他禾本类植物.全世界的竹亚科植物约有119属,已被描述的1 500个种是森林植被极为重要的组成部分[1].竹子以根状茎繁殖为主,且很少开花结实,利用花和果实等生殖性状对竹子进行分类受到极大限制.现阶段系统划分主要依据竹子的营养器官如地下茎、杆箨、分枝方式,但这些形态性状极为相似,难以观察和判定,且容易受到生境、生长阶段、生长部位乃至个体大小等因素的影响,加上在后期栽植中出现不同程度的无性系变异特征被放大,造成分类界定上的混乱.事实上,一些形态上有显著差异的竹子,可能有更大程度的遗传相似性[2],因而为解决由形态性征不同而造成的分类问题,直接从分子角度进行研究尤为重要.
核苷酸序列作为最直接、最稳定的标记性状,现已成功应用于植物各分类阶元的系统发育构建,其最大优势在于能提供相对客观的分子证据,可以直接反映物种的遗传变异,是近年来竹子系统鉴定的主要手段之一.竹亚科各类分类研究涵盖的范围较广,不同学者先后以族或属(群)为主题作了相应的研究和探讨.研究主要涉及倭竹族[3]、刚竹属Phyllostachys[4]、空竹属Cephalostachyum[5]等. DNA条形码(DNA barcoding)技术是近年来在物种鉴定领域发展的一种新方法[6].一段好的DNA条形码必须具有较高的种间变异性,以区别不同的物种,同时保持较低的种内变异性[7],核基因组的ITS是较为热门的一个DNA条形码[8-9].在第三届DNA条形码国际学术大会上,与会者一致建议选择进化速率较快的ITS探讨它们在陆生植物中的通用性、分辨率和适合程度,ITS适合于属种间较低等级的系统发育研究[10]. ITS是指18SrDNA和28SrDNA基因的间隔序列,由ITS1区、5.8S基因、ITS2区组成.内转录区ITS1和ITS2区作为非编码区,承受的选择压力小,变化相对较大,而核糖体DNA中的5.8S基因在大多数生物中趋于保守,物种间变化小,被广泛应用于系统分类鉴定的研究.虽然人们越来越多地应用ITS序列鉴定物种,但仍没有运用ITS序列鉴定竹亚科不同物种的研究.本研究利用GenBank公布的ITS序列及本试验测得的ITS序列进行比较分析,实现准确而快速的鉴定.
HTML
-
本文所用的ITS序列共76条,其中包括GenBank序列14条,试验所得的序列共62条.材料采摘于重庆市梁平县竹博园和重庆茶山竹海景区.材料为竹类植物的叶片,叶片采下后用硅胶保存(慈竹属:1种;刚竹属:37种;箬竹属:2种;唐竹属:2种;矢竹属:3种;大明竹属:1种;赤竹属:5种;巴山木竹属:1种;倭竹属:3种;短穗竹属:2种;簕竹属:2种;酸竹属:2种;少穗竹属:1种).拉丁名、属名依据中国植物志.
-
本次试验的具体步骤参照张汉尧等[10]的改良CTAB方法.取样品叶片0.05 g,加入2%PVP和2%巯基乙醇在研钵中用液氮磨碎,转入1.5 mL离心管中;加入900 μL 2×CTAB提取缓冲液,混匀后置于65 ℃水浴锅中保温1 h,其间轻轻摇动几次;冷却至室温,加入600 μL 24: 1的氯仿-异戊醇,颠倒混匀,12 000 r/min离心10 min;取上清液,加入2/3体积冰冷的异丙醇,轻微摇匀,-20 ℃放置30 min以上;12 000 r/min离心10 min,倒掉上清液;用70%乙醇洗沉淀2次,100%乙醇洗1次;室温下晾干DNA,20 min左右,加灭菌蒸馏水溶解,-4 ℃低温保存备用.
-
PCR扩增选用ITS引物,ITS上游引物ITS2的序列为5'-AGAAGTCGTAACAAGGTTTCCGTAGG-3',下游引物ITS4的序列为5'-TCCTCCGCTTATTGATATGC-5'[11].反应体系25 μL包括:PCRMax 12.5 μL;正反引物(10 mmoL/L)各1.0 μL;模板DNA 2 μL;用ddH2O补充至25 μL.反应条件:98 ℃预变性2 min;98 ℃变性10 s,55 ℃退火5 s,72 ℃延伸15 s,40个循环;最后72 ℃延伸5 min;-4 ℃低温保存.用1%琼脂糖电泳检测PCR扩增情况,得到的产物送上海华大科技有限责任公司进行序列纯化及正反双向测序.
-
试验所得的DNA序列与GenBank数据库中下载的序列利用Clustal X进行对比,并辅以人工校正,去除引物序列及测序质量较差的序列,得到可供分析的450 bp长度的ITS序列;然后利用MEGA6.06分析不同竹种序列间的变异频率,计算各样品的种内、种间距离;最后采用K2P模型进行遗传距离分析并构建NJ(Neighbor-Joining)距离树[12],利用Bootstrap(1 000次重复)检验各分支的支持率.
2.1. DNA的提取
2.2. PCR扩增及测序
2.3. 数据处理
-
竹亚科各竹种序列经Clustal X比对后长度为450 bp,分析其各样品ITS序列长度、GC含量(表 1).
-
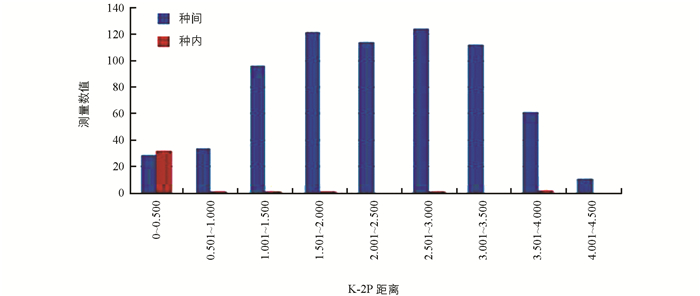
基于K2P模型计算遗传距离,对1-76号样品进行分组,见表 1.结果显示,种内遗传距离为0~3.907,平均遗传距离为0.370;种间遗传距离为0~4.394,平均遗传距离为2.287.种间变异明显大于种内变异,符合条形码种间变异大、种内变异小的要求,证明ITS序列能够准确区分竹亚科的不同竹种(图 1).
-
利用MEGA6.06分析软件的K-2P距离构建NJ(Neighbor-Joining)距离树(图 2),拓扑结构的可靠性用1 000次重复的自展检验(Bootstrap analysis)来评估,分支支持率低于50%的略去.
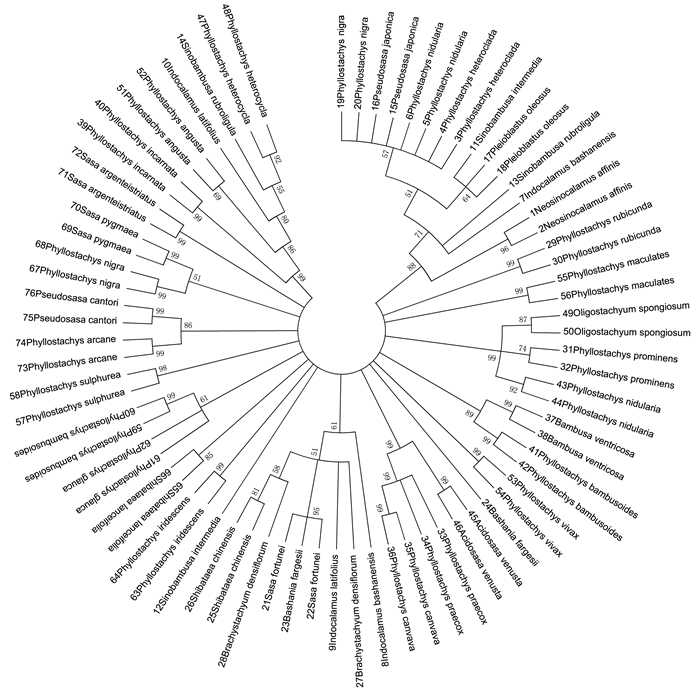
从NJ树可看出,慈竹Neosinocalamus affinis的序列(1,2)聚为一支,支持率为96%;鹅毛竹Shibataea chinensis的序列(25,26)聚为一支,支持率为81%;红后竹Phyllostachys rubicunda的序列(29,30)聚为一支,支持率为99%;髙节竹Phyllostachys prominens的序列(31,32)聚为一支,支持率为74%;安吉水胖竹Phyllostachys canvava的序列(35,36)聚为一支,支持率为99%;罗汉竹Bambusa ventricosa的序列(37,38)聚为一支,支持率为99%;红壳雷竹Phyllostachys incarnata的序列(39,40)聚为一支,支持率为99%;黄槽斑竹Phyllostachys bambusoides的序列(41,42)聚为一支,支持率为99%;实肚竹Phyllostachys nidularia的序列(43,44)聚为一支,支持率为92%;黎竹Acidosasa venusta的序列(45,46)聚为一支,支持率为99%;金丝毛竹Phyllostachys heterocycla的序列(47,48)聚为一支,支持率为92%;斗竹Oligostachyum spongiosum的序列(49,50)聚为一支,支持率为87%;黄古竹Phyllostachys angusta的序列(51,52)聚为一支,支持率为69%;黄竿乌哺鸡竹Phyllostachys vivax的序列(53,54)聚为一支,支持率为99%;斑苦竹Phyllostachys maculates的序列(55,56)聚为一支,支持率为99%;金竹Phyllostachys sulphurea的序列(57,58)聚为一支,支持率为98%;寿竹Phyllostachys bambusoides的序列(59,60)聚为一支,支持率为99%;淡竹红哺鸡竹Phyllostachys iridescens的序列(63,64)聚为一支,支持率为99%;狭叶倭竹Shibataea lanceifolia的序列(65,66)聚为一支,支持率为85%;紫竹Phyllostachys nigra的序列(67,68)聚为一支,支持率为99%;无毛翠竹Sasa pygmaea的序列(69,70)聚为一支,支持率为99%;铺地竹Sasa argenteistriatus的序列(71,72)聚为一支,支持率为99%;黄槽石绿竹Phyllostachys arcana的序列(73,74)聚为一支,支持率为99%;托竹Pseudosasa cantori的序列(75,76)聚为一支,支持率为99%.水竹Phyllostachys heteroclada的序列(3,4)、篌竹Phyllostachys nidularia的序列(5,6)、曙筋矢竹Pseudosasa japonica的序列(15,16)与毛金竹Phyllostachys nigra的序列(19,20)聚为一支,支持率为57%;菲白竹Sasa fortunei的序列(21,22)与巴山木竹Bashania fargesii(23)聚为一支,支持率为64%.早竹Phyllostachys praecox的序列(33,34)与安吉水胖竹(35,36)聚为一支,支持率为99%.但巴山箬竹Indocalamus bashanensis(7,8)、阔叶箬竹Indocalamus latifolius (9,10)、晾衫竹Sinobambusa intermedia(11,12)、红舌唐竹Sinobambusa rubroligula (13,14)、秋竹Pleioblastus oleosus(17,18)、巴山木竹Bashania fargesii(23,24)、短穗竹Brachystachyum densiflorum(27,28)没有聚为一支,种内遗传距离较远.
3.1. 序列分析
3.2. 种内、种间变异分析
3.3. 聚类分析
-
DNA条形码技术从分子水平对物种进行鉴定,具有准确性高、高通量、操作简便等特点,避免了形态学上的误差,是现代生物分类的发展方向之一.理想的DNA条形码应该具备明显的种间差异,同时种内差异要足够小,并且能够使用单引物对其进行扩增,通过双向测序得到高质量的序列[11].在竹亚科的研究中常见的核DNA片段主要是ITS,ITS以点突变为主,变异位点相互独立,适合于种或种下水平的分析,更重要的是通过两旁rDNA保守序列上的通用引物,可对很广范围的种类进行PCR扩增和测序,作为物种分类的通用标记[12],该核片段的应化适度提高了一些竹种间系统发育关系的分辨力.本研究结果表明,核基因ITS序列具有较好的PCR扩增效率和测序成功率,同时具有较大的种间变异和较小的种内变异,在样本量较大的情况下,其物种鉴定成功率达到81.6%,故ITS序列可用于竹类资源的物种鉴定研究.但研究表明这些核基因在竹子中均不是单拷贝[13]. Song等[14]经过分析发现,一些竹种的ITS序列出现假基因化,致使拷贝间的序列差异明显,影响了系统发育关系的正确构建.另外,杂种存在也是分类混乱不可忽视的一个原因,因为竹类植物具有其特殊的生理与生态特性[12],在已探讨的竹类杂交亲和力试验中,同属不同种的竹子间杂交亲和力较强,部分不同属但生态特性相似的竹子间杂交亲和力也较强[15].竹类植物很少开花,由于其特殊的分布方式和繁殖方法,这些杂交竹种在外观上很难分辨,仅靠营养器官不足以对其进行准确地分辨,对于竹亚科这样一类经历了快速分化、序列分化水平程度低的类群,可能仍然需要结合大量核基因序列进行佐证[16].研究证明,通过増加DNA序列的取样量可提高对类群关系的分辨力[17].
基于DNA序列差异的分析被认为是最直接的阐明类群间系统关系的方法.但由于竹子过长的世代周期和快速的物种分化导致一些种属内序列分化程度不高,试验难以得到竹类植物族间、属间一致的演化关系和良好的拓扑支持率,值得进一步研究.
 DownLoad:
DownLoad: