




-
随着微生物组研究的深入,越来越多的研究表明多细胞动物不单单只是一个简单的生物体,而是携带了众多微生物体的“超级生物体”. 2019年5月,Nature杂志宣布了人类微生物组计划第二阶段的完成,该阶段主要包括了微生物与早产、微生物与炎症性肠病、微生物组与糖尿病三部分研究内容[1].与人类微生物组研究一样,昆虫肠道微生物的研究同样也取得了非常瞩目的成果[2-7].研究证明同种昆虫的肠道微生物种类和丰度会因食物、地域和生长发育等不同而出现差异[8-10],相同地域相同食物的不同昆虫之间肠道微生物也会存在较大差异[11].肠道微生物功能的研究证明,肠道微生物与昆虫的生长发育、生殖系统及消化系统发育、运动能力、迁徙聚集等有着密切的关系.果蝇肠道中的植物乳酸杆菌被证实可通过调节胰岛素信号通路促进宿主黑腹果蝇的生长速率[12-13],产气荚膜梭菌、东方醋酸杆菌等细菌同样对果蝇的生长发育有促进作用[14-15].有研究证明肠道微生物通过改变肠道微环境的氧化还原反应通路促进昆虫中肠组织的发育[16-17];而醋酸杆菌属细菌可促进果蝇卵巢组织中乙醛脱氢酶的合成,乙醛脱氢酶进一步促进果蝇的卵巢发育并提高产卵量[18].群聚、迁徙行为对于某些昆虫种群的生存是至关重要的,如蝗虫、蜜蜂、蟑螂,研究表明昆虫的群聚、迁徙行为会受到肠道微生物影响.在农业害虫蝗虫的肠道中,肠道微生物可合成分泌大量的愈创木酚和少量的苯酚,两种物质均可促进蝗虫的群聚[19];在被微孢子虫感染后,蝗虫肠道微环境稳态失衡会严重影响蝗虫的群聚[20].通过蟑螂肠道微生物的研究发现,蟑螂肠道微生物群产生的挥发性羧酸同样对蟑螂的群聚行为至关重要[21].
草地贪夜蛾(Spodoptera frugiperda)是世界重大迁飞性农林害虫,主要以禾本科植物(玉米、高粱、水稻)为食,危害巨大.自2019年1月从东南亚进入我国云南、广西地区,随着温度升高和农作物的丰富,草地贪夜蛾随季风快速迁徙至全国19个省份[22-25].虽然美国和巴西开展了草地贪夜蛾肠道优势细菌分离鉴定工作[26-27],但在我国暂未见相关的研究报道.本研究采集了重庆巫山、巫溪地区玉米田中的草地贪夜蛾样本,通过传统培养法以及16S rDNA测序对草地贪夜蛾的肠道优势菌进行了初步分离鉴定,以期为后续的深入研究奠定基础.
HTML
-
草地贪夜蛾幼虫采自重庆巫溪及巫山的玉米田中.
-
马铃薯琼脂培养基(PDA):马铃薯200.0 g,葡萄糖20.0 g,琼脂15.0 g,加蒸馏水至1 000 mL.
改良LB培养基(LBG):胰蛋白胨10.0 g,酵母提取物5.0 g,NaCl 10.0 g,葡萄糖5.0 g,琼脂15.0 g,加蒸馏水至1 000 mL.
牛肉膏蛋白胨培养基(NA):牛肉膏5.0 g,蛋白胨10.0 g,NaCl 5.0 g,琼脂15.0 g,加蒸馏水至1 000 mL.
-
微生物裂解直接PCR试剂盒(Takara);PCR扩增引物27F::5'-AGAGTTTGATCCTGGCTCAG-3';1492R:5'-GGTTACCTTGTTACGACTT-3',由生物工程(上海)股份有限公司合成;1×Taq MasterMix(purple),北京博迈德基因技术有限公司.
-
在超净台中,将活的草地贪夜蛾幼虫浸泡于75%的乙醇中5 min,进行表面消毒.表面消毒完成后,取出中肠置于灭菌的1.5 mL EP管中,加1 mL灭菌的磷酸缓冲液(PBS)后充分震荡,以此为原液备用.
-
取100 μL制备的肠道原液于新的1.5 mL EP管中,加入900 μL灭菌PBS配制成10-1组,递次稀释为10-2和10-3备用.各取10-2组和10-3组溶液100 μL涂布于不同的固体培养基上,在37 ℃的培养箱中培养24 h.随机挑选单菌落,在分离培养基上进行单斑划线获得纯培养,并染色观察.
-
1) 取50 μL裂解缓冲液于灭菌的1.5 mL EP管中.
2) 用灭菌的牙签从平板上挑取单菌落,置于EP管中搅动几下后取出.
3) 80 ℃热变性15 min后,低速离心,取1~5 μL裂解后的上清液作为PCR反应的模板.
4) 用细菌通用引物27F和1492R对上述模板进行PCR,扩增细菌16S rDNA序列.反应体系(50 μL)如下:模板5 μL,引物27F/1492R各1 μL,1 ×TaqMasterMix(purple)43 μL.反应程序为:96 ℃预变性10 min;96 ℃变性30 s,55 ℃退火30 s,72 ℃延伸1 min 30 s,35个循环;72 ℃延伸10 min.将扩增产物送至华大基因科技股份有限公司测序.
5) 将获得的16S rDNA基因序列在GenBank(http://blast.ncbi.nlm.nih.gov/Blast.cgi)与数据库已登录基因序列进行同源比对,下载GenBank中同源性较高序列,利用MEGA5.0软件,使用N-J法进行1000次步长计算,构建系统发育树.同时将获得的16S rDNA基因序列在RDP数据的Classifer程序(https://rdp.cme.msu.edu/classifier/classifier.jsp)进行比对,获得同源性较高序列的相关信息.
1.1. 样本材料
1.2. 实验用的培养基
1.3. 主要试剂
1.4. 中肠内容物提取
1.5. 肠道细菌分离
1.6. 分离菌株的初步鉴定
-
采用PDA培养基、LBG培养基和NA培养基,分别从巫山、巫溪地区采集的草地贪夜蛾幼虫肠道中各分离获得15个分离株.通过细菌通用引物27F/1492R对30个分离株进行PCR扩增测序获得其16S rDNA序列.将所有测序结果利用Blastclust进行同源性聚类分析,同源性大于97%的聚为统一单元,共获得6个聚类单元(表 1).
将测序结果分别在GenBank和RDP两个数据库比对获得相关信息.比对结果显示,本次从入侵巫山地区的草地贪夜蛾肠道分离的15个分离株中,WS1,WS2,WS6,WS11,WS14和WS17等6个分离株属于克雷伯氏菌属(Klebsiella);WS15,WS3,WS4和WS18属于假单胞菌属(Pseudomonas);WS7,WS5,WS9和WS12属于不动杆菌属(Acinetobacter);WS8属于肠杆菌属(Enterobacter).巫溪地区的15个分离株中,WX1,WX2,WX3,WX4,WX5,WX6,WX7,WX8,WX9,WX10,WX11,WX12和WX19属于克雷伯氏菌属(Klebsiella);WX21属于不动杆菌属(Acinetobacter);WX17属于气单胞菌属(Aeromonas).不动杆菌属的分离株在聚类分析中分为两个聚类单元,WS7,WS5和WS9聚类到OTU3,WS12和WX21聚类到OTU4.在所有分离株中,克雷伯氏菌属(Klebsiella)的丰度最高,占63%;其中假单胞菌属(Pseudomonas)和肠杆菌属(Enterobacter)仅在巫山地区分离得到,气单胞菌属(Aeromonas)仅在巫溪地区分离得到(表 1).
-
为进一步了解不同地区草地贪夜蛾肠道优势菌的遗传多样性,对30个肠道微生物分离株进行了系统发育分析.对分离株16S rDNA序列进行BLAST比对分析,采用MEGA5.0软件N-J法构建系统发育树(图 1)[28].
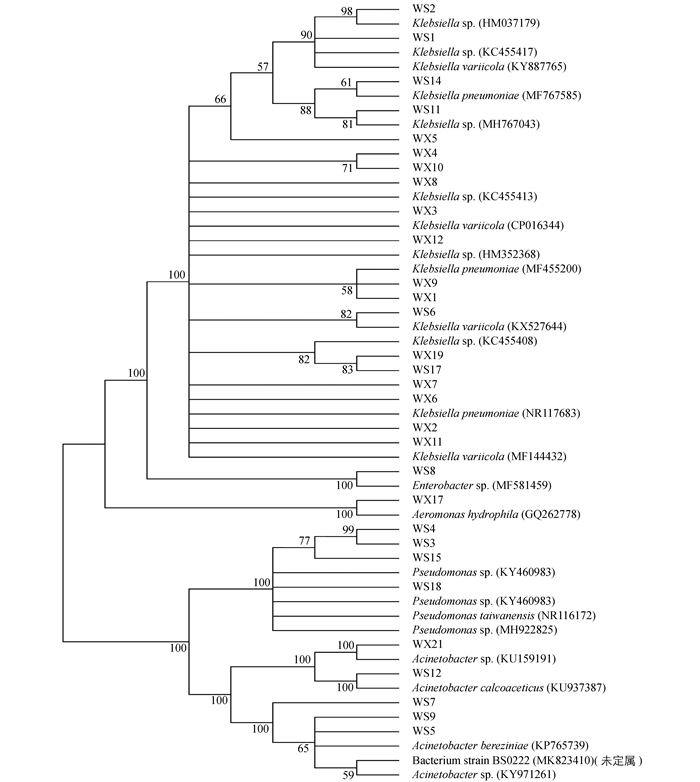
由图 1可见,30个分离株在进化发育树分属于5个属,分别是克雷伯氏菌属(Klebsiella)、不动杆菌属(Acinetobacter)、假单胞菌属(Pseudomonas)、气单胞菌属(Aeromonas)及肠杆菌属(Enterobacter).其中19株属于克雷伯氏菌属(Klebsiella),占整体的63%,但这些菌株在种间存在一定的差异,即种间多样性较大. WS7,WS5,WS9,WS12和WX21都属于不动杆菌属(Acinetobacter),但聚类为OTU3的WS7,WS5和WS9与聚类到OTU4的WS12和WX21在进化关系上存在一定的差异.值得关注的是假单胞菌属(Pseudomonas)虽然在巫山分离株中占有率较高,但在巫溪分离株中没有分离得到,这有可能是地区环境差异导致的.
2.1. 培养法分离鉴定草地贪夜蛾肠道细菌
2.2. 肠道细菌的系统发育树分析
-
肠道微生物对宿主的生活习性、生长发育及生理状态等方面有着至关重要的作用.研究发现,对常规农药具有抗性的草地贪夜蛾肠道微生物中,存在着大量的可降解农药的细菌[26];此外,草地贪夜蛾肠道中的细菌可以调节植物的防御反应以及促进幼虫的生长发育[27, 29].
本研究通过纯培养的方法,分离培养并初步鉴定了入侵重庆巫山、巫溪地区的草地贪夜蛾肠道微生物中的优势细菌,共分离鉴定到了5个属的细菌,分别为克雷伯氏菌属(Klebsiella)、假单胞菌属(Pseudomonas)、不动杆菌属(Acinetobacter)、肠杆菌属(Enterobacter)、气单胞菌属(Aeromonas).其中克雷伯氏菌属(Klebsiella)的比例最高,且种水平多样性较高,推测该区域草地贪夜蛾肠道微环境适于克雷伯氏菌属(Klebsiella)的定植,从而使克雷伯氏菌属(Klebsiella)在肠道微环境中大量增殖.此外,两个地区之间存在地区差异的分离株,假单胞菌属(Pseudomonas)和不动杆菌属(Acinetobacter)仅分离于巫山地区害虫样品,气单胞菌属(Aeromonas)仅分离于巫溪地区样本,推测是草地贪夜蛾在适应不同地区生态环境过程产生的差异.
巴西学者Luis Gustavo de Almeida等从对农药具有抗性的草地贪夜蛾肠道中分离到了以下8个属的细菌:肠球菌属(Enterococcus)、代尔夫特菌属(Delftia)、勒克氏菌属(Leclercia)、细杆菌属(Microbacterium)、假单胞菌属(Pseudomonas)、节细菌属(Arthrobacter)和葡萄球菌属(Staphylococcus)[26];Flor Acevedo等从美国宾夕法尼亚州的草地贪夜蛾肠道中分离获得了泛菌属(Pantoea)、肠杆菌属(Enterobacter)、拉恩氏菌属(Rahnella)、拉乌尔菌属(Raoutella)和克雷伯氏菌属(Klebsiella)5个属的细菌[27].与巴西和美国两项研究相比,重庆地区与美国宾州地区草地贪夜蛾肠道微生物中细菌在属一级的水平存在一定的重合性,推测可能是因为同一物种肠道微环境具有一定保守性导致的;但与巴西地区差异较大,可能与巴西地区高强度的农药选择压力有关.
综上,我们首次在国内报道了重庆地区玉米田中草地贪夜蛾的肠道细菌分离株,与美国和巴西的研究结果存在不同程度的差异,说明其组成受地理生态环境、宿主生理状态等多方面的影响.这些肠道微生物是否也会影响宿主的抗性、生长发育、迁飞,不同细菌之间的相互关系怎样,仍有待进一步研究.
 DownLoad:
DownLoad: