




-
DNA宏条形码(DNA metabarcoding)技术是一种以DNA条形码技术与下一代测序技术为基础、可用于分析混合样品DNA以获得群类物种多样性的方法[1-3]. DNA条形码技术是利用一段标准的DNA条形码序列来对物种进行鉴定与分类,由加拿大动物学家Hebert最早提出[4].传统的sanger测序仅能对单一样品进行DNA测序,而高通量测序技术可对混合DNA进行同时测序,可获得大量具有生物学信息的DNA片段,进行物种多样性研究及组分分析[5].随着DNA条形码与测序技术的不断进步以及条形码数据库的逐步完善,DNA宏条形码技术现已成为现代生物学研究热点,广泛应用于生物多样性研究[6-8].
DNA宏条形码自创立以来,已被应用于诸多领域. Farruggia等利用此技术检测放牧食草动物粪便,分析其膳食结构及植物多样性[9];宋飏等利用COI基因分析了太白山土壤动物多样性[10];Taberlet等利用高山草甸土壤样品分析了植物多样性,并为开展生物多样性调查的全面标准化提供了可能性[11].此外,DNA宏条形码技术还应用于食性分析[12]、古生物研究[13]、生态评估[14]等领域.
莼菜(Brasenia schreberi)是睡莲科水生草本植物,富含蛋白质、膳食纤维和多糖物质,具有降血脂、降血糖、抗氧化和抗癌等多种生理活性[15-16].前人研究结果表明,土壤微生物多样性将直接影响到作物的正常生长[17].本文选取ITS(Internal Transcribed Spacer)基因序列作为分子标记,应用DNA宏条形码技术对莼菜大田不同种植年限的土壤真菌多样性进行研究,为莼菜资源的保护及科学利用提供数据支持.
HTML
-
2017年5月上旬,采集重庆市石柱县冷水镇八龙村种植年限分别为1年、5年、10年、15年、20年的莼菜大田土壤,每种生境选取5 m×5 m的3个样方,采用五点取样法在样方内随机采取3份土样,取样深度(0~25) cm.土样上层与下层的比例相同,取样后干冰运输.采集样地如表 1所示.
-
使用MoBio强力土壤微生物DNA提取试剂盒(北京昊成铭泰科技有限公司)进行各样品的总DNA提取,得到15份总DNA.将相同生境的3份DNA进行等浓度混合,得到后续PCR的5份总DNA.
-
PCR反应体系为25 μL,含dNTP Mixture(大连宝生物公司,2.5 mmol/L)2 μL、PCR buffer(大连宝生物公司,10×)2.5 μL、正反向引物(2.5 μmol/L)各1.0 μL、Taq DNA聚合酶(大连宝生物公司)1 μL、总DNA约1 μL(30~50 ng),其余用ddH2O补至25 μL.
PCR扩增引物ITS1(大约250 bp),正向引物序列为GGAAGTAAAAGTCGTAACAAGG,反向引物序列为GCTGCGTTCTTCATCGATGC. PCR的循环程序为:94 ℃预变性1 min;94 ℃变性5 min,退火温度55 ℃持续1 min,72 ℃延伸1 min,30个循环;最后72 ℃延伸7 min.
每份总DNA扩增3次,然后将3份PCR产物等浓度混合. PCR产物的测序使用Illumina MiSeq平台,采用250PE测序策略,由上海派森诺生物科技有限公司完成.
-
采用滑动窗口法对FASTQ的双端序列逐一做质量筛查,利用FLASH软件进行配对连接,通过QIIME软件调用USEARCH检查并剔除模糊碱基和嵌合体序列,从而获得每个样本的有效序列.
-
试验用QIIME软件,调用UCLUST对前述序列按97%的序列相似度进行OUT聚类,选取每个OTU中最长序列为代表序列;利用QIIME中的BLAST,将代表序列与UNITE数据库比对,获得OUT分类学信息.
首先进行OTU划分并对分类地位鉴定结果进行统计,再用Excel和Mothur软件做目水平上的群落组成图和样品间VENN图.
-
使用R软件vegan程序包对各样本的Chao1丰富度指数、ACE丰富度指数、Shannon多样性指数和Shannon-Wiener多样性指数进行计算.
-
使用GraPhlAn,对样本总体在各分类水平的组成构建等级树,同时以不同颜色区分各分类单元,并通过节点大小反映它们的丰度分布.
使用R软件中的pheatmap程序包,对丰度前50位的属进行聚类分析并绘制热图.
1.1. 样品采集与准备
1.2. 样品总DNA提取
1.3. PCR扩增及测序
1.4. 数据处理与分析
1.4.1. 原始数据整理、过滤及质量评估
1.4.2. OTU划分和分类地位鉴定
1.4.3. Alpha多样性分析
1.4.4. 分类学组成分析
-
通常,每1个OTU被视为1个微生物物种,试验对OTU在门、纲、目、科、属、种各分类水平进行了鉴定.结果表明,随着种植年限的增加,OTU数呈先增加后减少的规律,说明莼菜土壤真菌数量先增后减,在10年生莼菜土壤中达到峰值(表 2).
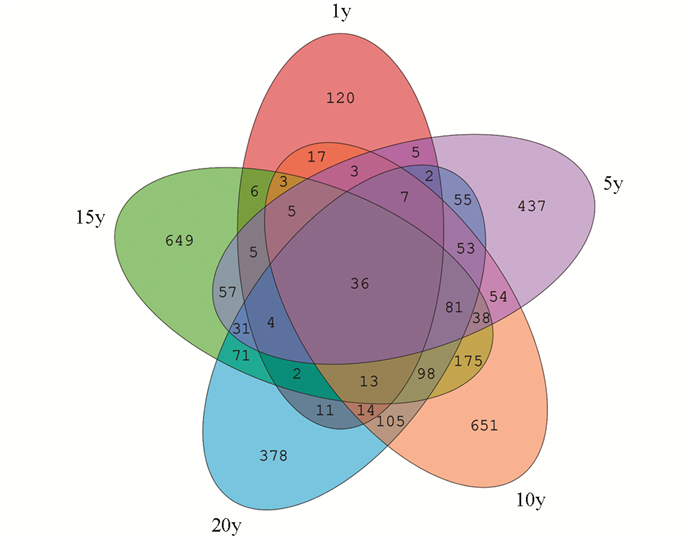
通过共有OTU的Venn图,发现5个样本共有的OTU数为36,1年生样本独有OTU数为120,5年生样本独有OTU数为437,10年生样本独有OTU数为651,15年生样本独有OTU数为649,20年生样本独有OTU数为378.可以看出样本之间的相似性和差异性(图 1).
-
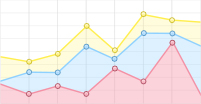
各莼菜土壤真菌群落中,Chao1丰富度估计指数从小到大依次为1年生、5年生、20年生、15年生、10年生;ACE丰富度估计指数大小次序和Chal指数相同;Simpson多样性指数和Shannon多样性指数从小到大依次均为1年生、20年生、5年生、15年生、10年生.试验结果看出,莼菜土壤真菌的群落丰富度和多样性指数均呈先增后减的趋势(表 3).
-
通过OTU划分和分类鉴定,1年生莼菜真菌群落由7门、18纲、45目、70科、83属、138种构成;5年生莼菜真菌群落由7门、21纲、54目、95科、136属、244种构成;10年生莼菜真菌群落由7门、22纲、61目、110科、173属、295种构成;15年生莼菜真菌群落由7门、19纲、57目、87科、119属、210种构成;20年生莼菜真菌群落由7门、22纲、52目、94科、136属、217种构成(表 4).不同年限莼菜土壤真菌在科、属、种水平上呈现出不同的群落组成,但均在10年生莼菜土壤中数量最多.
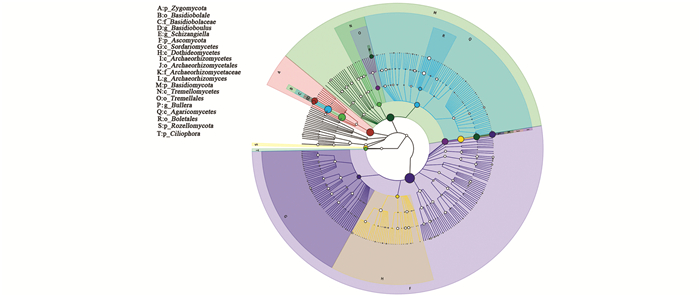
基于GraPhlAn的样本总体分类等级树图能快速发现优势微生物类群.在属的水平上蛙粪霉属、裂梗霉属、Archaeorhizomyces属、酵母属为优势属.蛙粪霉属在样本中的相对丰度随着生长年限的增加而增加;裂梗霉属有随着生长年限增加含量增加的趋势,但在15年生样本中含量低于10年生样本. Archaeorhizomyces属真菌主要集中在1年生莼菜土壤中,相对丰度达到67.2%,在其他样本中含量较低;酵母属在1年生样本中含量较低,15年生样本中含量较高,在其他3个样本中含量基本一致(图 2).
通过聚类,可以反映样本之间的群落组成相似度. 图 3中,红色代表在对应样本中丰度较高的属,绿色代表丰度较低的属.由图 3可知,10年生莼菜土壤与15生莼菜土壤首先聚类,然后与20年生莼菜土壤聚类,然后与5年生莼菜土壤聚类,最后与1年生莼菜土壤聚类.

2.1. OTU分类学鉴定
2.2. Alpha多样性分析
2.3. 不同年限莼菜土壤真菌群落的组成
-
研究发现,随着种植年限的增加,莼菜土壤真菌的chao1指数、ACE指数、simpson指数、Shannon-Wiener指数均呈现先增加后减少的趋势,都在第10年达到峰值.纳小凡等发现宁夏枸杞土壤微生物多样性与种植年限有密切关系[17].胡凯等研究发现桉树土壤真菌数量年际变化显著,随着种植年限的增加呈明显下降趋势[18].谢光新等在对茶树的土壤真菌群落研究中发现,10年生茶园根际真菌数量比2年生茶园多[19],说明土壤真菌群落的变化受所生长的植株的影响而成差异性变化.通过对门、纲、目、科、属各个水平的分析,发现不同年限真菌的变化呈现一定的规律.不同年限各种真菌的数量呈现不同变化,有的升高,有的降低,有的则维持相对稳定状态.因为影响土壤真菌数量的因素不只年限的变化,还有土壤因子、生态扰动等作用[20-21].在后续研究中,应加入其他因素对莼菜大田真菌群落进行综合评价.
DNA宏条形码技术相比传统测序单一物种鉴定,可同时对混合样本进行测序分析,具有省时省力的优点,现已成为物种多样性分析的利器[22-23].研究者认为DNA宏条形码技术利用序列数的多少来表示物种个体数量的多少,两者之间变化趋势一致,但没有完美契合,因此DNA宏条形码技术做定性分析的结果是可靠的,但定量分析仍需进一步验证[24-25].随着高通量测序技术的不断发展和分析方法的进一步完善,DNA宏条形码技术将成为现代生态学物种多样性研究的有效措施和必然趋势.
 DownLoad:
DownLoad: