


 下载:
下载:
-
开放科学(资源服务)标识码(OSID):

-
苹果是世界上重要的经济作物,我国是苹果的重要生产国,栽培面积和产量均位居全球首位.苹果花叶病是我国苹果生产栽培中最常见的病毒性病害,能够引起花叶、斑驳、坏死等症状[1].感病叶片栅栏组织细胞排列松散,细胞器畸变,液泡非正常增生,叶绿体变成不规则的球形,膜结构遭到破坏,严重影响光合能力,最终导致果实产量和品质下降,对苹果产业健康发展危害较大[2].
2017年,日本研究者利用高通量测序(Next Generation Sequencing,NGS)技术从表现花叶和坏死症状的苹果叶片中鉴定出一种新的病毒,命名为苹果坏死花叶病毒(apple necrotic mosaic virus,ApNMV)[3].ApNMV属于雀麦花叶病毒科(Bromoviridae)等轴不稳环斑病毒属(Ilarvirus),基因组结构为三分体,包括RNA1,RNA2和RNA3.RNA1和RNA2均为单顺反子,分别编码与病毒复制相关的蛋白MET/HEL和RNA依赖的RNA聚合酶POL,前者蛋白的C端与后者蛋白的N端在这两个蛋白互作中起主要作用[3-5].RNA3为双顺反子,分别编码一个5'端的运动蛋白(movement protein,MP)和一个3'端的外壳蛋白(coat protein,CP),CP中含有α-螺旋、β-折叠、锌指结构域和RNA结合区域,在病毒的复制、蛋白翻译及与寄主的互作中发挥重要功能[2-4].已有的研究表明,ApNMV在我国苹果花叶病样品中的检出率高达92%,且在我国苹果种植区普遍发生,是引起我国苹果花叶病的重要病原[2-4, 6-8].韩国[9]、印度[10]也在少量苹果花叶病样品中检测出ApNMV.除苹果外,沙果(Malus spp.)[6]、海红果(Malus micromalus Makino)[7]和山楂(Crataegus spp.)[11]也被鉴定出是ApNMV的自然寄主.这些结果说明了ApNMV可能具有较宽的自然寄主范围,值得引起重视,而新检测技术的开发也有助于新寄主的发现.
目前,NGS测序技术和逆转录-聚合酶链式反应(reverse transcription-polymerase chain reaction,RT-PCR)是检测ApNMV的主要技术手段[3-4, 9, 11].尽管RT-PCR技术具有检测灵敏、快速等优势,但受到试验条件、检测设备和成本的限制.NGS虽然具有检测灵敏度强、检测范围广、效率高的特点,但检测过程耗时费力,成本较高,难应用于口岸或田间ApNMV的快速检测和鉴定,且不适于对大量样品的检测.血清学方法尤其是酶联免疫吸附分析方法(enzyme-linked immunosorbent assay,ELISA)因其能够对大量样品进行快速、灵敏、准确的检测,以及试验成本低、方法简单等优点,成为植物病毒鉴定、分类以及检测的重要工具.近年来,果树病毒的原核表达及抗血清制备已得到广泛应用[12-14].但是,目前国内外尚无ApNMV血清制备及有效检测的相关报道.因此,本研究利用分子生物学手段在大肠杆菌中高效表达ApNMV的CP,制备多克隆抗血清,建立ApNMV血清学常规检测体系,对于生产中该病害的诊断和防控具有重要意义.
全文HTML
-
ApNMV带毒和健康的苹果叶片样品由本实验室保存.
原核表达载体pET-28a(+)由本实验室保存,多糖多酚植物总RNA提取试剂盒和大肠杆菌感受态细胞DH5α购自天根生化科技有限公司(天根,北京),限制性内切酶和T4 DNA连接酶购自宝日医生物技术有限公司(TaKaRa,北京),反转录试剂dNTP Mix,M-MLV reverse transcriptase,RNase inhibitor购自于普洛麦格生物技术有限公司(Promega,北京),高保真酶Phusion high-fidelity DNA polymerase购自美国赛默飞世尔科技公司,2×Taq Mix购自北京冰达生物科技有限公司,DNA凝胶回收试剂盒和质粒提取试剂盒购自美国Axygen公司,大肠杆菌原核表达菌株BL21(DE3)购自北京博迈德生物技术有限公司,羊抗兔酶标二抗购自北京博尔迈生物技术有限公司,化学发光显色液和PVDF膜购自Millipore公司(美国).
-
叶片总RNA提取使用多糖多酚植物总RNA提取试剂盒(天根),具体操作方法按说明书进行.反转录合成cDNA,体系为:dNTP Mix(10 mmol)1 μL,5×M-MLV buffer 4 μL,M-MLV reverse transcriptase(200 U·μL-1,PROMEGA)0.5 μL,RNase inhibitor(40 U/μL,PROMEGA)0.5 μL,随机六聚引物(10 μM)0.5 μL,Oligo(dT)18引物(10 μM)0.5 μL,用灭菌ddH2O补足至20 μL.混匀后,37 ℃温育1 h.之后,进行PCR扩增目的基因,体系为:cDNA 1 μL,dNTP Mix(10 mmol)0.4 μL,5×Phusion HF buffer 4 μL,上游引物ApnCP-F(CGC GGA TCC ATG GTG TGC AAT CGC TGT CA下划线序列为Bam H Ⅰ酶切位点)0.8 μL,下游引物ApnCP-R(CCG AAG CTT GAC ATC CAA AAG GTC TTC ATC G下划线序列为Hind Ⅲ酶切位点)0.8 μL,Phusion DNA polymerase(2 U·μL-1,Thermo Scientific)0.25 μL,用灭菌ddH2O补足体系至20 μL.PCR反应循环参数:98 ℃预变性30 s,98 ℃变性10 s,65 ℃退火30 s,72 ℃延伸30 s,循环32次;72 ℃再延伸10 min,4 ℃保存,预期目的片段大小为675 bp.PCR产物经1.5%琼脂糖凝胶电泳、成像分析后,使用AxyPrep DNA凝胶回收试剂盒纯化回收目的片段.ApNMV的检测参考Xing等[4]方法.
-
分别将回收产物和原核表达载体pET-28a(+)用Bam H Ⅰ,Hind Ⅲ进行双酶切.体系为:回收产物或pET-28a(+)质粒30 μL,Bam H Ⅰ,Hind Ⅲ各2.5 μL,10×K buffer 5 μL,用灭菌ddH2O补足体系至50 μL;混匀后,37 ℃温育15 h,加5 μL 10×loading buffer终止酶切反应,酶切产物经1%琼脂糖凝胶电泳、成像后,切下目的条带,使用凝胶回收试剂盒纯化回收.分别取4 μL双酶切后的目的基因回收产物和pET-28a(+)回收产物、1 μL 10×T4 DNA buffer和1 μL T4 DNA Ligase(TaKaRa)进行4 ℃过夜连接反应.将连接液转化入50 μL DH5α感受态细胞,涂板(卡那抗性),37 ℃过夜培养后,筛选阳性克隆,送生工生物工程(上海)股份有限公司测序.
-
将基因序列、读框均正确的重组质粒及pET-28a(+)分别转化大肠杆菌表达菌株BL21(DE3),37 ℃过夜培养,挑取单菌落,过夜培养,保存甘油菌并将过夜菌液按1∶20比例接种到新鲜卡那抗性LB培养基,小量表达.小量表达成功后大量表达,接种100 μL甘油菌到50 mL卡那抗性LB培养基中过夜培养,再按1∶20比例转接种到1 000 mL培养基中,37 ℃,160 r/min,振荡培养;当OD600达到0.6左右时,向培养的菌液中加入0.5 mmol IPTG,诱导蛋白表达,37 ℃,160 r/min,振荡培养2 h;4 ℃,8 000 r/min,20 min离心回收菌体.纯化蛋白用Ni Sepharose 6 Fast Flow,最后回收的蛋白集中在250 mmol咪唑/8M Urea PBS中,用于后续多抗免疫.
-
小量表达时,离心收集菌体,加入1/10体积样品缓冲液,震荡悬浮,100 ℃煮沸5 min,用12%的胶进行SDS-PAGE分析,浓缩胶电压为8 V/cm,分离胶为15 V/cm.考马斯亮蓝R-250染色1 h后,室温摇床上脱色3 h.表达产物的Western blot分析参考Towbin等[15]的方法,以保存的His-tag抗体为一抗,羊抗兔抗体(博尔迈,北京)为酶标二抗,最后用TMB显色.大量表达检测则使用经过纯化后的蛋白为上样样品,其余同小量表达SDS-PAGE及Western blot分析.
-
先在试验兔耳静脉取血2 mL制备少量正常血清,作为阴性对照.之后,向经过纯化的蛋白产物中加入等体积的福氏完全佐剂(首次)或不完全佐剂(后续)进行乳化,使用2只无特定病原体(specific pathogen free,SPF)级新西兰兔在屏障环境下进行皮内多点注射免疫.每周五进行免疫,第一次免疫后休息一周,共免疫6次.完成免疫后,采血2 mL进行ELISA效价检测,如效价不够需追加免疫,如效价达标,则进行全采血及ELISA检测.制备的抗血清经过滤加入0.09%的叠氮化钠,4 ℃保存,备用.另外,将所得抗血清进行5倍间隔梯度稀释,以表达的目的融合蛋白为抗原包被,利用间接ELISA检测方法测定血清的效价.
-
参考施曼玲[16]、郑世玲[17]的方法,以制备的稀释400倍后的ApNMV抗血清为一抗,购买的羊抗兔抗体(博尔迈,北京)为酶标二抗,间接ELISA(ID-ELISA)法检测田间苹果叶片样品.以不加抗原为空白对照进行校零,测定样品在波长为450 nm处的吸光值(OD值),根据临界值=待测样品OD值/阴性对照OD值进行结果的判断.本研究中,阴性对照OD450 nm数据为3个健康苹果样品的均值,待测样品OD450 nm测定均做3个重复;P/N值为待测样品与对照样品OD450 nm比值,若P/N值>2,则认为样品带毒,否则不带毒.
1.1. 材料
1.2. 方法
1.2.1. 植物总RNA提取及RT-PCR
1.2.2. 原核表达重组质粒构建
1.2.3. CP基因诱导表达及蛋白纯化
1.2.4. 表达产物的SDS-PAGE及Western blot检测分析
1.2.5. 抗血清的制备及效价评定
1.2.6. 间接ELISA检测苹果叶片样品
-
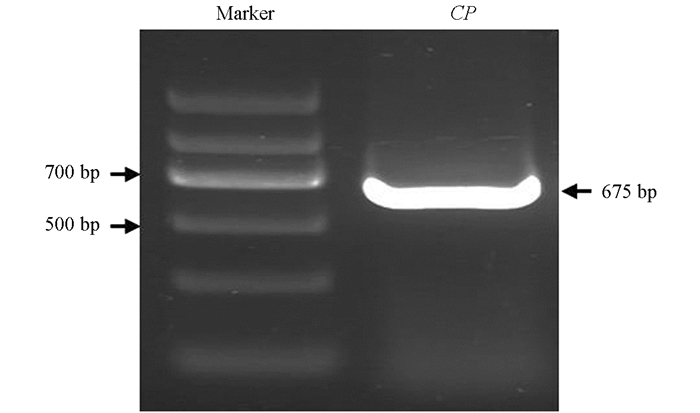
提取苹果花叶病叶片样品总RNA,经反转录后,用特异性引物ApnCP-F/R进行RT-PCR扩增,产物经1.5%琼脂糖凝胶电泳检测,结果显示,两个泳道中均有明亮、单一且符合预期目的基因大小(675 bp)的特异性条带(图 1).
切胶纯化目的条带,Bam H Ⅰ和Hind Ⅲ双酶切后,连入经相同限制性内切酶处理后的原核表达载体pET-28a(+),连接后得到重组质粒.经过测序验证,表明CP基因以正确的序列和读码框连接到表达载体中.在NCBI分别进行Blastn和Blastx比对,发现获得的ApNMV CP序列在核苷酸水平和蛋白水平与已登陆的序列相似性均为96%~100%,其中,所得序列与ApNMV分离物AM75 RNA3(登录号:KY808387)编码的CP序列完全一致.表明CP序列较为保守,适合用于蛋白表达制备抗血清用以检测ApNMV.
-
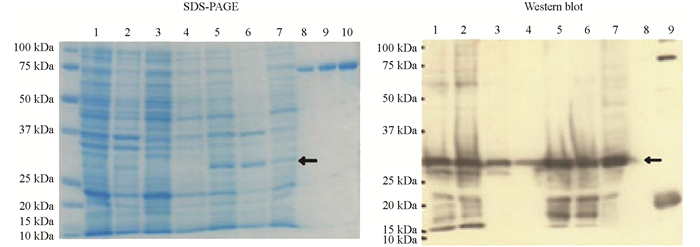
37 ℃过夜培养大肠杆菌表达菌株后,经煮沸裂解细胞,释放表达蛋白,对小量表达样品进行SDS-PAGE和Western blot检测分析.结果显示,裂解溶液、沉淀部分和上清部分均能在分子量约25 kDa处检测到与重组蛋白预期大小一致的目的条带;SDS-PAGE分析显示,未经IPTG诱导的泳道中则无明显的符合预期大小的条带,但Western blot能够杂交出微弱条带;经IPTG诱导表达后,重组蛋白表达量显著增加(图 2).由此表明,CP基因被成功诱导表达.
-
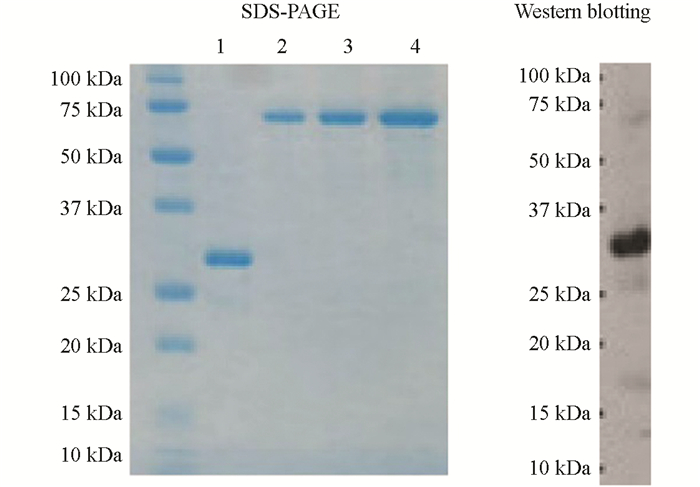
小量蛋白表达分析证明了目的蛋白被成功表达,进一步扩大培养,诱导蛋白大量表达后,对大肠杆菌裂解后的沉淀部分用Ni Sepharose 6 Fast Flow进行蛋白纯化,经过回收的蛋白集中在250 mmol咪唑/8M Urea PBS中,浓度1.2 mg/mL,体积2 mL,共纯化得到重组蛋白2.4 mg.另外,将得到的纯化蛋白进行SDS-PAGE和Western blot检测分析,在分子量约25 kDa附近有主要目的蛋白条带出现,说明试验纯化出符合要求的目的蛋白(图 3).
-
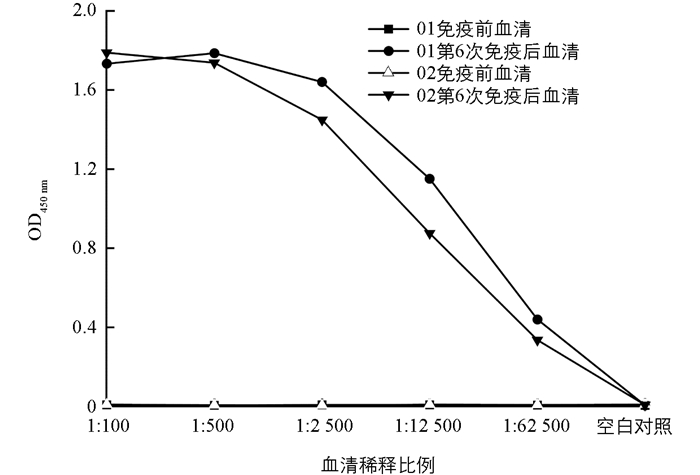
将纯化的目的蛋白免疫2只SPF级新西兰兔,最后获得了ApNMV的特异性抗血清.以表达的目的融合蛋白为抗原包被,利用间接ELISA检测方法测定抗血清的效价.结果表明,2只兔子的采血血清稀释62 500倍后仍能表现明显的阳性反应,同时与免疫前血清、空白对照没有明显的血清学反应(图 4).
-
利用实验室制备的ApNMV抗血清,通过间接ELISA法对来自田间的苹果叶片进行检测.样品的检测结果显示,样品1~6的P/N值均大于2,判断为ApNMV阳性;7~9样品P/N值均小于2,判断为ApNMV阴性(表 1).该ELISA检测结果与前期RT-PCR检测结果一致,表明制备的ApNMV抗血清能够应用于苹果叶片ApNMV的检测.
2.1. CP基因克隆及重组质粒构建
2.2. CP基因诱导表达检测
2.3. 重组蛋白大量表达及纯化
2.4. 抗血清的制备和效价评定
2.5. 间接ELISA法检测苹果叶片样品
-
ApNMV是一种新的植物病毒,是导致我国苹果花叶病的重要病原物[2-4, 6, 8].考虑到苹果栽培在我国农业生产中的重要地位,加强对该病毒的检测并进一步提出合理科学的防控措施尤为必要.血清学检测是病毒检测的重要手段,尤适用于大量样品的检测,被广泛用于田间病毒病的发生分布调查.病毒特异性抗血清的制备是进行血清学检测的基础,血清通过与相应抗原的特异结合发生免疫反应,选择合适的血清反应方法不仅可以检测到寄主植物中病毒存在与否,同样能够进行病毒含量多少的测定[18].
等轴不稳环斑病毒属病毒的CP由源于RNA3的亚基因组RNA4编码,3'端能够形成复杂的二级结构,参与病毒的复制,激活病毒的侵染过程,具有较强的保守性[19-21].为此,本研究利用较强保守性的外壳蛋白为表达目的蛋白,制备出多克隆抗血清,以此避免检测过程中假阴性结果的出现.同时,由于构建的重组质粒表达产物中不含有植物蛋白成分,在用血清学方法检测相应病毒病原时不易出现假阳性[22].
研究表明,插入目的基因、载体及菌株类型与培养条件等因素可能会影响外源基因的表达[23].本研究选用了高效表达载体pET-28a(+)作为构建ApNMV CP基因原核表达的骨架载体,该载体采用T7 lacI原核启动子,能够极大提高翻译效率.该载体携带6×His标签,一方面有助于利用His抗体杂交目的融合蛋白,借助蛋白分子量标准,判断目的基因表达情况;另一方面,所表达出的融合蛋白能够利用Ni柱进行快速纯化.此外,6×His标签蛋白分子量小,免疫原性低,有助于与目的蛋白连接时减少对其空间构象及活性的影响.构建成功的重组载体导入到大肠杆菌表达菌株中进行融合蛋白的诱导表达,本研究首先使用小量表达,以证明构建的载体能够成功表达出目的蛋白;再扩大培养菌株,大量表达以纯化出足够量的目的蛋白,以此免疫兔子获得较高浓度针对ApNMV CP的特异性抗体.进行抗血清效价评定时,先利用纯化出的融合蛋白作为抗原,测试显示制备的抗血清效价较高.进一步以此抗血清为基础,利用间接ELISA方法对田间采集的苹果叶片样品进行ApNMV检测,检测结果与RT-PCR结果相一致,表明制备出的抗血清灵敏度高.考虑到苹果花叶病毒(apple mosaic virus,ApMV)、李属坏死环斑病毒(prunus necrotic ringspot virus,PNRSV)与ApNMV较近的遗传进化关系[4, 8],本研究试图利用ApMV,PNRSV阳性苹果叶片样品评估制备的抗血清的特异性.但多年来,本实验室未从苹果中检测到这两种病毒,因而利用感染了PNRSV的桃叶片作为待测样品,结果显示本研究制备的ApNMV抗血清与桃中PNRSV未见明显的免疫反应,说明该抗血清具有较好的特异性.
-
本研究成功进行了ApNMV CP基因的原核表达,制备出抗血清,并首次建立了一种简便、快速、灵敏度高、特异性良好的间接ELISA检测方法,为ApNMV田间调查提供了可行的检测手段,为病害诊断与防控提供了技术基础.