


 下载:
下载:
-
光动力治疗(PDT)是利用光敏剂,在一定波长的光照射下,引发光化学反应,产生单线态氧,进而破坏肿瘤,达到治疗肿瘤的目的.目前,第一代光敏剂——血卟啉,因组分复杂、肿瘤靶向性不足、作用深度较浅等缺点,逐渐被第二代光敏剂——替莫卟吩(m-THPC)所取代[1-4].与血卟啉相比,m-THPC具有较高的细胞毒性、肿瘤选择性和较深的作用深度等优点,但存在水溶性不足、肿瘤靶向性差、光毒性等缺点.由于肿瘤组织的代谢异常,导致大部分肿瘤细胞的叶酸受体高表达,且叶酸受体与叶酸具有较高的亲和性[5-7].基于上述原因,前期研究中将羧酸卟啉与叶酸偶联,成功制备了叶酸-羧酸卟啉偶联物,细胞实验发现,与羧酸卟啉相比,宫颈癌细胞(Hela细胞)对叶酸-羧酸卟啉的摄取是羧酸卟啉的35倍,且具有较强的光毒性和较低的暗毒性[8].但叶酸-羧酸卟啉的水溶性不足和卟啉的光动力活性较低,限制其广泛应用.因此,本研究利用叶酸、羧酸卟吩和聚乙二醇二氨(NH2-PEG-NH2),以酰胺键偶联,制备了一种叶酸-PEG-羧酸卟吩(光敏剂7),其分子式为
光敏剂7在水中的溶解度为40.1 mg/L,细胞靶向性和毒性也远高于羧酸卟吩.然而,光敏剂7仍存在相对分子质量较大、组分不定、光敏剂化学结构改变等问题,导致光动力活性受到影响[9].
正常组织细胞内谷胱甘肽(GSH)的浓度是细胞外液中的200倍以上,且肿瘤细胞内的GSH是正常细胞的7~10倍,导致肿瘤细胞内具有较强的还原性.因此,还原敏感型药物递送系统成为人们研究的热点.还原敏感型药物主要通过二硫键与GSH的巯基进行可逆转换而实现[10-11].多肽具有合成简单、对肿瘤有被动靶向、多肽修饰的光敏剂能改善油-水分配系数等优点[12-13].因此,本研究采用叶酸受体介导的叶酸靶向策略,m-THPC作为光敏剂,引入穿膜肽八聚精氨酸(R8)连接基团,同时以二硫键为化学键,成功制备了还原敏感型叶酸-多肽-m-THPC(光敏剂1),并对光敏剂1的生物活性进行探究.
全文HTML
-
主要仪器:Varian 640型红外光谱仪(美国Varian公司);DU800型紫外可见分光光度计(美国Beckman公司);Mercuryplus-400 MHZ核磁共振仪(美国Varian公司);Agilent 1200液相色谱仪(美国Agilent公司);KDH 150B红光治疗仪(输出波长600~700 nm,北京科电微波电子有限公司);AXIMA Resonance Lcms 2010质谱仪(日本岛津公司).
主要试剂:9-芴甲氧羰基-(三苯甲基)半胱氨酸王树脂(Fmoc-Cys(Trt)-王树脂)、9-芴甲氧羰基-精氨酸8-COOH(Fmoc-(Arg)8-COOH)、六氟磷酸苯并三氮唑-1-氧基三吡咯烷基磷(PyBop)、N,N-二异丙基乙基胺(DIPEA)和1-羟基苯并三氮唑(HOBT)均购于上海吉尔生化有限公司;二甲亚砜、吡啶、N,N-二甲基甲酰胺(DMF)、三氟乙酸(TFA)、三乙胺、碳酸三氯甲基酯(三光气)、三异丙基硅烷(TIS)、1,2-乙二硫醇(EDT)和肼均购于中国国药集团;叶酸(FA)(Sigma公司,美国),反相硅胶(ODS-AQ,YMC公司,日本),透析袋(MWCO=1 000,生工生物,上海).二甲亚砜、吡啶和DMF在使用前均经减压蒸馏处理;其余试剂均为AR,使用前未经处理.
-
色谱柱为300 extend C18柱(5 μm,4.6×150 mm);流动相A为5%乙腈-水溶液(含0.1%三氟乙酸),流动相B为乙腈(含0.1%三氟乙酸),梯度洗脱,在15 min内流动相A从100%到0%,流速:1 mL/min;柱温:25 ℃;进样量:10 μL;紫外可见吸收检测器检测波长:410 nm.
-
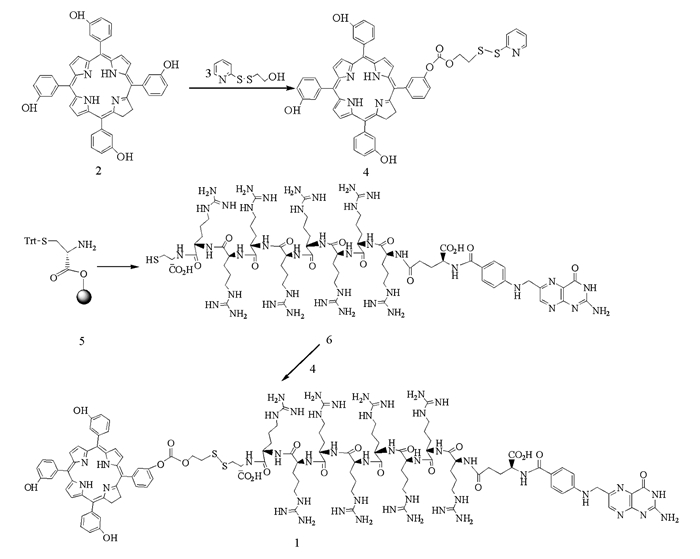
在氩气保护下,将90.62 mg(0.48 mmol)2-(2-羟基-乙二硫)吡啶[14]、0.067 mL(0.48 mmol)三乙胺和47.93 mg(0.48 mmol)三光气溶于15 mL二氯甲烷中,室温反应10 min后,滴加至含有300 mg(0.44 mmol)m-THPC和0.067 mL(0.48 mmol)三乙胺的15 mL在乙腈中,室温反应6 h,减压蒸干溶剂,反相硅胶(ODS-AQ,甲醇)柱色谱分离,得到60.43 mg化合物4,产率为15.36%. UV-Vis (CH3OH):414,514,540,594和648 nm;1HNMR (500 MHz,DMSO-d6):δ9.81,8.72~8.21 (m,6H,CH,Pyrrole),8.21~7.06 (m,20H),4.48 (s,2H,-CH2-,-OCH2-),4.17 (s,4H,-CH2-,Pyrrole),3.23 (s,2H,-CH2-,-CH2S-),1.23 (s,2H,-NH-);MS (ESI):894.1722 (M + H);HPLC:96.6%.
-
采用多肽固相合成法,在氩气保护下,将1.0 g Fmoc-Cys(Trt)-王树脂置于接肽瓶中,加入2 mL哌啶DMF(20%),振荡1 h,减压抽滤,重复3次,用DMF(3 mL × 3)和异丙醇(3 mL × 3)冲洗树脂,得H2N-Cys(Trt)-王树脂.称取1.19 g Fmoc-(Arg)8-COOH(0.8 mmol)溶于5 mL DMF,加入1.04 g PyBOP(2 mmol)、0.27 g HOBT(2 mmol)和0.41 g DIPEA(3.2 mmol),在氩气保护下,室温反应30 min,加入H2N-Cys(Trt)-王树脂,室温反应4 h,用DMF(5 mL × 3)和异丙醇(5 mL × 3)冲洗树脂,减压抽干,加20%哌啶DMF(2 mL × 3),振荡1 h,减压抽滤,得H2N-(Arg)8-Cys(Trt)-王树脂.将0.35 g叶酸(0.8 mmol)溶于5 mL混合溶剂(n(DMF):n(DMSO)=1:3),加入1.04 g PyBOP(2 mmol)、0.27 g HOBT(2 mmol)和0.41 g DIPEA(3.2 mmol),在氩气保护下,室温活化30 min,加入H2N-(Arg)8-Cys(Trt)-王树脂,室温反应12 h.用DMF(5 mL × 3)和DCM(5 mL× 3)冲洗树脂,减压抽干,得FA-(Arg)8-Cys(Trt)-王树脂.将FA-(Arg)8-Cys(Trt)-王树脂置于接肽瓶中,在氩气保护下,加入2%肼DMF(2 mL × 3),室温反应10 min,减压抽干,用DMF(5 mL × 3)和异丙醇(5 mL × 3)冲洗树脂,减压抽干.加入4 mL混合溶剂(n(TFA):n(H2O):n(TIPS):n(EDT)=92.5:2.5:2.5:2.5),反应30 min,重复3次,收集滤液,真空干燥,得到FA-(Arg)8-Cys-SH粗品.反相硅胶柱色谱分离(40%乙腈水),得到0.53 g纯品. MS(MALDI-TOF):m/z 1793.96;HPLC:98.6%.
-
光敏剂1的合成路线见图 1.在氩气保护下,将50 mg化合物6(0.0278 mmol)溶于5 mL水中,用饱和Na2CO3溶液调节pH值至6.8,将该溶液加至溶有24.9 mg化合物4(0.0278 mmol)的5 mL DMSO中,室温反应2 h,透析,冷冻干燥,反相硅胶柱色谱分离(45%乙腈水),得到8.64 mg光敏剂1,产率为12.06%. 1HNMR (500 MHz,DMSO-d6) δ:8.72(s,β-H,Pyrrole),8.24~8.39(s,2H,ArH),8.03~8.09(m,4H,ArH),7.41~7.52(m,ArH),4.54(m,-CH2-,Arg),4.19~4.24(m,-CH2-,Arg),3.50(t,-CH2-,Arg),1.73~1.76;MS(MALDI-TOF):m/z 2575.18;HPLC:99.1%.
-
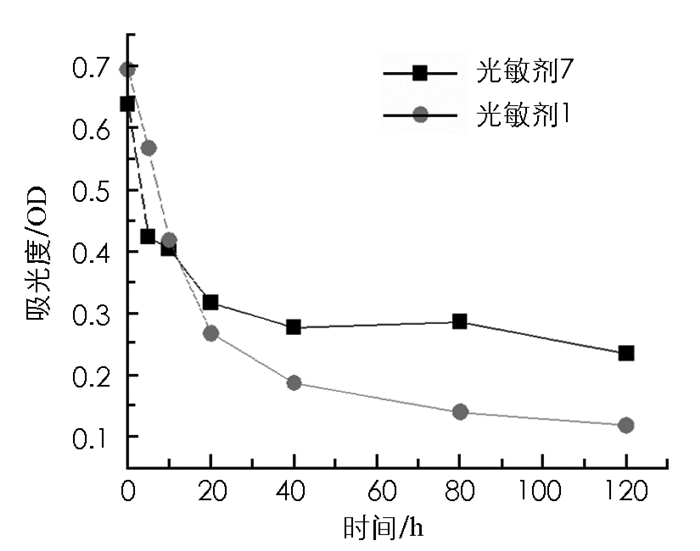
将还原敏感型光敏剂1和光敏剂7溶于10 mmol/L的磷酸盐缓冲液(PBS,pH=7.0),配制成浓度为12 μmol/L的溶液,在暗室中,用红光治疗仪在600~700 nm下光照射,分别于0,5,10,20,40,80,120 min取样,测定吸光度.光稳定性的计算公式为:ASoret (120 min)/ASoret (0 min) × 100%.
将过量的还原敏感型光敏剂1和m-THPC分别溶于1 mL水中,振荡2 h,涡旋10 min,保证溶解完全,在转速为12 000 r/min条件下离心40 min,取上清液,冷冻干燥,准确称量,获得溶解度.
-
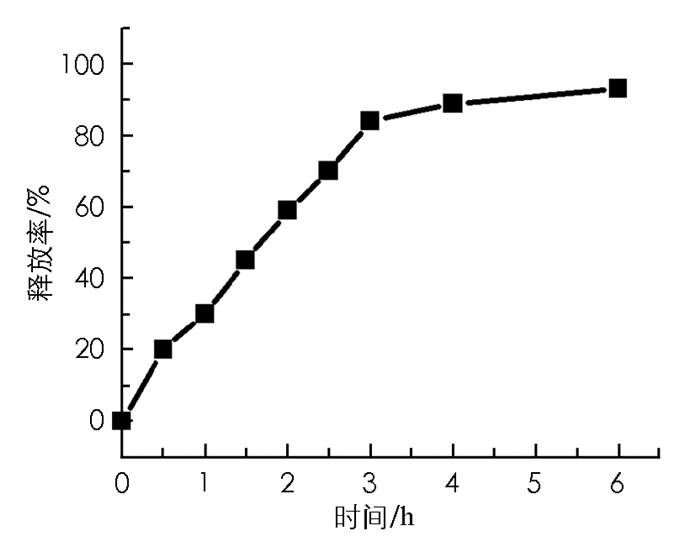
将还原敏感型光敏剂1(0.01 mmol,25.77 mg)溶于PBS(10 mL)中,配制成浓度为1 mmol/L的溶液,迅速加入5倍GSH(0.05mmol,15.37mg),在37 ℃涡旋混匀,分别于0,0.5,1.0,1.5,2.0,2.5,3.0,4.0和6.0 h取样,离心,取上清液,微孔滤膜过滤后,供HPLC分析.
-
将浓度为5 × 104个/mL的人宫颈癌HeLa细胞株(叶酸受体阳性细胞)和人肺腺癌A549细胞(叶酸受体阴性细胞)分别接种于6孔板上,在无叶酸的1640(10%胎牛血清)培养24 h,各设3个试验组.试验组1为在孔中加入还原敏感型光敏剂1,终浓度为16.5 μmol/L;试验组2为在孔中加入光敏剂7,终浓度为16.5 μmol/L;试验组3为同时在孔中加入还原敏感型光敏剂1和FA,其中靶向光敏剂的终浓度为16.5 μmol/L,叶酸的终浓度为3 mmol/L.各实验组培养24 h后弃培养液,DPBS洗涤3次,每孔用4%多聚甲醛固定30 min,吸出液体,DPBS洗涤,甘油封片,激光共聚焦测定各孔细胞中光敏剂的荧光强度(Ex:480 nm;Em:660 nm).
-
于96孔培养板中接种浓度为5 × 104/mL的HeLa细胞2孔,各分15个组,培养至对数生长期,除对照组外,各组分别加入浓度为0.5,0.9,1.9,3.8,7.5,15.0,30.0,60.0和120.0 μmol/L的光敏剂1和7,培养24 h,冷PBS洗3次,加新鲜培养液,分别用红光治疗仪照射5 min.光照后继续培养24 h,加含MTT(5 g/L)的PBS溶液20 μL,培养4 h,弃去培养液,加入100 μL DMSO,振荡10 min,用酶标仪测定吸光值(570 nm),计算细胞存活率:SR =实验组OD值/对照组OD值× 100%.
1.1. 主要仪器与试剂
1.2. 高效液相色谱条件
1.3. 还原敏感型叶酸-多肽-m-THPC光敏剂1的合成
1.3.1. 化合物4的合成
1.3.2. 化合物6的合成
1.3.3. 光敏剂1的合成
1.4. 测定还原敏感型光敏剂1的生物活性
1.4.1. 光稳定性和溶解性
1.4.2. 药物释放研究
1.4.3. 肿瘤靶向性测定
1.4.4. 细胞毒性研究
-
在中间体化合物4的制备中,采用2-(2-羟基-乙二硫)吡啶作为双硫键的引入基团,在三光气和三乙胺的条件下,得到的产率为15.36%.产率较低的原因在于,三光气活性较高,得到的产物不仅有一取代,还有二取代和三取代的副产物;为提高产率,可采用缓慢滴加的方式,将活化后的2-(2-羟基-乙二硫)吡啶,滴加至m-THPC溶液中.
光敏剂1的制备,是利用双硫键可以发生交换反应,但该反应为可逆反应,为获得较高的产率,应控制反应pH值在6~7之间.在弱酸性条件下,生成副产物巯基吡啶,会成盐,进而降低与光敏剂1发生巯基交换反应的可能性.因此,通过对反应pH值的控制,可以实现多肽修饰的还原敏感型叶酸靶向光敏剂1的制备.
-
以水为溶剂,测得光敏剂1的溶解度为13.32 mmol/L;m-THPC溶解后,离心,取上清液,UV-vis光谱分析,未见明显吸收峰,说明m-THPC在水中几乎不溶解.因此,八聚精氨酸的引入可显著改善光敏剂在水中的溶解度.
图 2为光敏剂1和光敏剂7的光稳定性实验结果.结果表明,在5 min内,光敏剂1和光敏剂7的稳定性分别下降18.39%和33.56%;光照10 min后,光敏剂1和光敏剂7的稳定性分别下降36.67%和39.70%.光照大于10 min,光敏剂1的稳定性小于光敏剂7.
-
在PBS(pH=7.0)、37 ℃和GSH作用下,还原敏感型光敏剂1可释放出母体药物m-THPC,6 h后释放率大于80%(图 3).
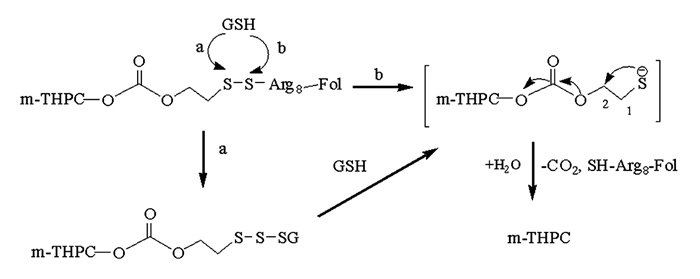
通过对光敏剂1的化学结构分析和文献调研,光敏剂1释放母体药物m-THPC的机理见图 4,其释放属于1,2-消除机理[15-16].
-
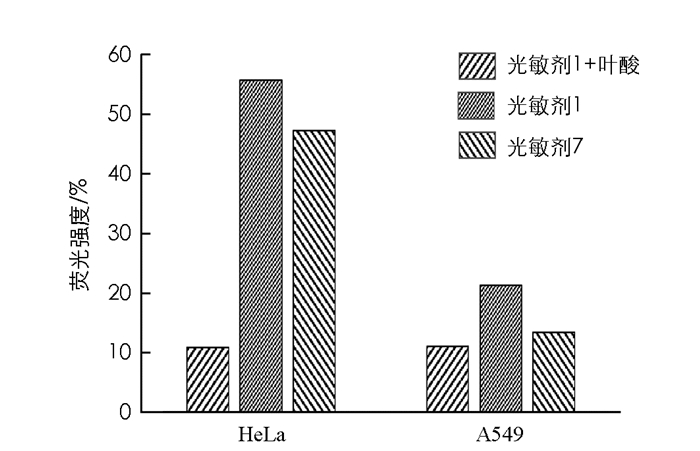
如图 5所示,在叶酸受体阳性细胞(HeLa)中光敏剂1和光敏剂7的荧光强度明显大于在叶酸受体阴性细胞(A549)中的荧光强度,即叶酸受体阳性细胞对光敏剂的摄取明显强于叶酸受体阴性细胞,且光敏剂1的吸收作用被大量加入的自由叶酸所抑制,说明光敏剂1的摄取是由肿瘤细胞表面的叶酸受体介导的内吞作用.光敏剂7的摄取是叶酸受体介导的内吞作用已在前期研究中被证实.
HeLa细胞和A549细胞中,光敏剂1的荧光强度大于光敏剂7的荧光强度,说明肿瘤细胞对光敏剂1的吸收高于光敏剂7.八聚精氨酸(R8)已被证实是一种良好的肿瘤细胞穿膜肽,光敏剂1中引入R8,可提高光敏剂1的肿瘤细胞靶向作用.
-
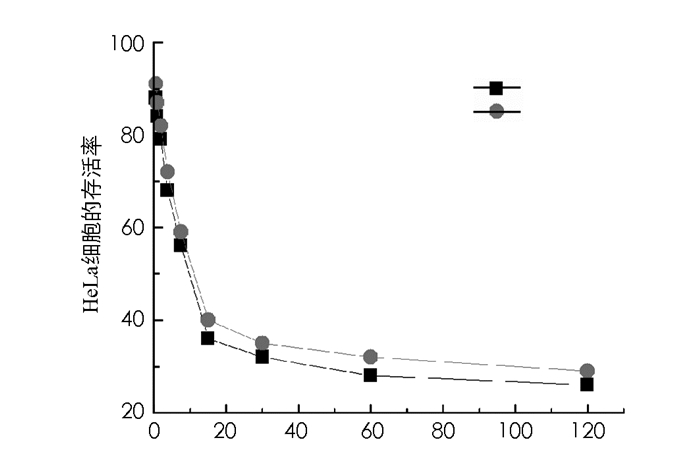
图 6为光敏剂1和光敏剂7的细胞毒性随浓度变化的实验结果.结果表明,在浓度为0.5~120μmol/L时,光敏剂1和光敏剂7的细胞毒性随浓度的增加而增加.当浓度为15μmol/L时,光敏剂1和光敏剂7对HeLa细胞的存活率分别降低至36.1%和39.7%.由于穿膜肽R8的靶向作用,导致光敏剂1的摄取大于光敏剂7,进而光敏剂1的细胞毒性大于光敏剂7.
2.1. 光敏剂1的制备
2.2. 光稳定性和溶解性
2.3. 药物释放
2.4. 肿瘤靶向性
2.5. 细胞毒性
-
采用八聚精氨酸(R8)为连接基团,叶酸为靶向基团,m-THPC为光敏剂,通过形成双硫键,制备了一种多肽修饰的还原敏感型光敏剂.研究发现,光敏剂1在水中的溶解度为13.32 mmol/L;在GSH作用下,6 h内光敏剂1释放率大于80%,文献分析发现,释放机理属于1,2-消除机理.细胞靶向性实验表明,穿膜肽R8的引入对肿瘤细胞的摄取具有协同作用,与光敏剂7相比,光敏剂1的细胞摄取率大于光敏剂7.细胞毒性实验证明,当光敏剂1浓度为15 μmol/L时,光敏剂1对HeLa细胞的存活率为36.1%,光敏剂7对HeLa细胞的存活率为39.7%,光敏剂1的细胞毒性高于光敏剂7.