


 下载:
下载:
-
开放科学(资源服务)标识码(OSID):

-
零维卤化物钙钛矿发光材料以其独特的量子限域效应以及可能存在的自陷激子行为逐渐成为近几年来光致发光领域的研究热点。光致发光主要包括激子直接复合发光、自陷态发光、缺陷态发光等机制。自陷态激子(Self-Trapped Exciton,STE)发光本质上源于晶格声子-激子相互作用,激子被“束缚”在晶格中,往往表现为宽光谱发射峰和大Stokes位移,在荧光粉[1]、闪烁体[2]和发光二极管[3]等方面具有重要的应用前景,近年来得到了广泛的关注。
本研究将基于零维发光材料Rb3BiCl6的实验研究结果,进一步探索其具体发光机理。该材料主要由[BiCl6]3-和零散分布的Rb+构成,晶格空间间隙较大,推测容易形成间隙和反位等本征缺陷。孤立的[BiCl6]3-八面体被Rb+隔开,因此电子和空穴往往被限制在某个八面体内部,而不在不同八面体之间迁移[4]。研究表明,Rb3BiCl6在295~345 nm处有1P1、3P2、3P1 3个激发峰,在420 nm处存在宽发射峰,显示出较大的Stokes位移,且具有软晶格特性和强激子-声子耦合效应,因而将其归结为自陷态发光机制[4]。然而在与此结构相似的Cs4PbBr6和Cs2ZrCl6中,都存在较易形成的本征缺陷,研究发现后者的Cl空位缺陷是深能级非辐射复合中心,对发光有猝灭作用[5-6]。而对于Rb3BiCl6,尽管实验已知其因强电子-声子耦合导致室温条件下不发光,但其本征缺陷的具体类型、能级位置,以及是否如同Cs2ZrCl6等容易形成卤素空位缺陷,仍需进一步探索。实验中并未观察到明显的缺陷发光峰,主要原因究竟是缺陷难以生成还是缺陷并不参与发光也有待讨论。在Cs2ZrCl6相关的研究中,也对其自陷态结构进行了探究,证明了不仅存在常见的单个[ZrCl6]2-内的局域自陷态,也存在相邻Cl捕获空穴形成Cl2-Vk(V表示空位)中心的Zr-Cl2-组合[7],说明类似的材料中可能存在多种不同的自陷态构象。另一种一维C4N2H14PbCl4发光材料的自陷态是[PbCl6]4-八面体单向轻微拉长的Jahn-Teller畸变[8]。尽管实验上已观测到Bi基零维钙钛矿中自陷激子发光的现象,但少有研究从微观原子尺度,特别是[BiCl6]3-八面体的结构畸变与动态弛豫模式出发,系统揭示其自陷态形成的理论机制。这种对晶格动力学与电子态耦合关系的理解缺失,限制了对该类材料发光性能的设计与调控。
因此本研究将通过理论计算,从缺陷态和自陷态两个方面具体探究Rb3BiCl6产生单一发射峰的原因。通过在可能的化学势取值范围内对可能产生的缺陷进行筛选,得到相对较易形成的缺陷。对相应缺陷进行相关带隙分析后,找到缺陷转变能级在禁带中的位置,从而确定这些缺陷均属于深能级缺陷,与实验中并未观测到缺陷发光这一现象相互验证。Rb3BiCl6的三重态结构优化结果显示某些[BiCl6]3-八面体体积约增加2%~4%,经进一步优化得到自限态结构,可见该类八面体发生轻微均匀膨胀,其能量比三重态自由态低约0.23 eV,处于自陷态发光的合理范围[4]。本研究为理解零维卤化物钙钛矿材料的发光机制提供了理论依据。
全文HTML
-
DFT计算采用广义梯度近似(Generalized Gradient Approximation,GGA)方法中的Perdew-Becke-Ernerhof(PBE)泛函。缺陷形成能的计算中,平面波截断能选取为500 eV,结构优化保持晶格参数和角度,原子相对位置同时可变(ISIF参数为3),倒易空间中的k点网格设置为2×1×3,以每个原子的Hellmann-Feynman力小于0.1 eV/nm、总能量变化小于10-5 eV作为结构优化的收敛标准。在激发态计算中,将原有晶胞扩胞成2×1×2的超胞,k点网格设置为1×1×1,其他参数与缺陷形成能相同。
在Rb3BiCl6中Bi作为重原子,自旋-轨道耦合(Spin-Orbit Coupling,SOC)作用过于明显,必须考虑能带劈裂现象所导致的能带变窄问题。同时,杂化泛函(Heyd-Scuseria-Ernzerhof 06,HSE06)的精确交换比过高会明显高估该体系的激发能、带隙等性质,若采用较低精确交换比的杂化泛函并考虑SOC作用又会极大增加计算成本。通常纯PBE泛函会低估体系的带隙和激发能[9-10],但经过测试和比较发现,纯PBE泛函的激发能和带隙计算值与考虑SOC作用的杂化泛函计算结果相近,考虑到两者产生的误差相消,因此所有与激发态相关的计算基本直接采用纯PBE泛函。
-
化合价为q价的X元素缺陷形成能可以定义为[11]:
式中:Etot[bulk]和Etot[Xq]分别是完美晶胞总能和缺陷晶胞总能; ni是i原子总数的变化,增加原子时ni为正值,反之则为负; μi是i元素化学势的大小,除Cl外所有原子的化学势均由最稳定条件下的固体单质和化合物确定,Cl的化学势则由实验合成时浓盐酸中Cl-的含量确定; EF是平衡费米能级的位置,因为缺陷的引入而有所改变。
X元素缺陷转变能级X是两种价态q1和q2形成能相同时对应的费米能级的位置,定义式为[1]:
其与导带底之间的间距决定了能级的深浅,影响发光性质。
-
固定超胞的总磁矩为2 μB,模拟三重激发态进行结构优化和性质计算,然后进行常规结构优化来得到自由态激子的构型和总能。根据自由态的[BiCl6]3-结构变化趋势,对其中一个[BiCl6]3-的相对位置进行特定方向微扰后获得自陷态的大致结构,并通过结构优化进一步确定。垂直发射能的计算采用ΔSCF方法[11],在固定原子结构的条件下,分别约束总磁矩为2 μB和0 μB,计算得到该结构下三重态和单重态基态的总能,两者的差值即垂直发射能。
1.1. 计算参数设置
1.2. 缺陷形成能和转变能级
1.3. 激发态计算
-
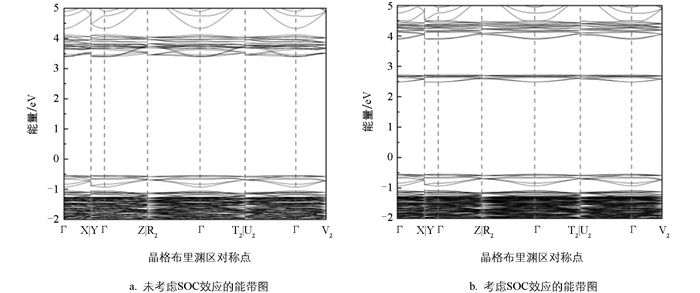
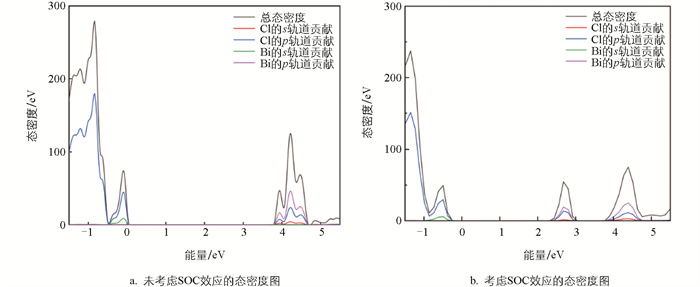
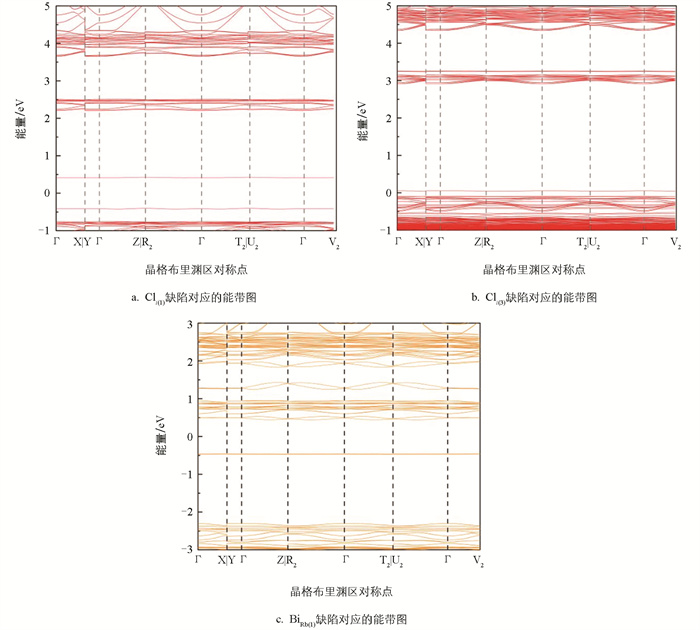
图 1给出了考虑和未考虑SOC效应的能带图,对比两组能带结构可见,考虑SOC效应的导带底显著下移,降幅约0.7 eV。这是由于Bi的6p轨道在SOC作用下发生劈裂并形成p1/2和p3/2两支,其中能量较低的p1/2构成了新的导带底。这种劈裂是重元素SOC效应的典型特征,与铅基卤化物钙钛矿(如MAPbI3)中观察到的现象类似。此外,考虑和未考虑SOC效应的缺陷能级价带顶和导带底的位置只发生了较小的变化,这意味着SOC作用对本研究体系缺陷能级与能带位置的绝对差值影响较小。图 2b中,能量大于2.4 eV处的导带底态密度图部分与图 2a相比也存在明显的劈裂,且Bi的p轨道贡献较大,这映证了能带图 1的结果。
与导带底的显著变化不同,价带顶的位置在考虑SOC效应后并未发生明显移动,这是由于价带顶位置主要由Cl的3p轨道贡献。Rb并未明显参与价带顶和导带底位置变化的贡献,这主要是由于Rb+分散地镶嵌在[BiCl6]3-八面体之间。
SOC效应是相对论量子力学中的一种重要效应,起源于电子自旋与其自身在原子核电场中运动所产生的磁矩之间的相互作用。在量子力学框架下,该相互作用的强度与原子序数的4次方成正比。因此在含有重元素(如Bi,Pb)的体系中,SOC效应极其显著地影响计算结果。在本研究中该效应表现为明显的能带劈裂。
-
Rb3BiCl6中3种元素的化学势取值范围必须满足:
式中:Δμi代表元素i的化学势相对于其最稳定单质相总能的差值,μBi和μRb近似等于最稳定态固体单质的总能EBi和ERb; ΔμBi、ΔμRb、ΔμCl分别是3种元素的化学势,皆为负值,以确保不会形成Bi、Rb、Cl的单质形态。ΔH为AxByCz的化合物形成焓,定义为[12]:
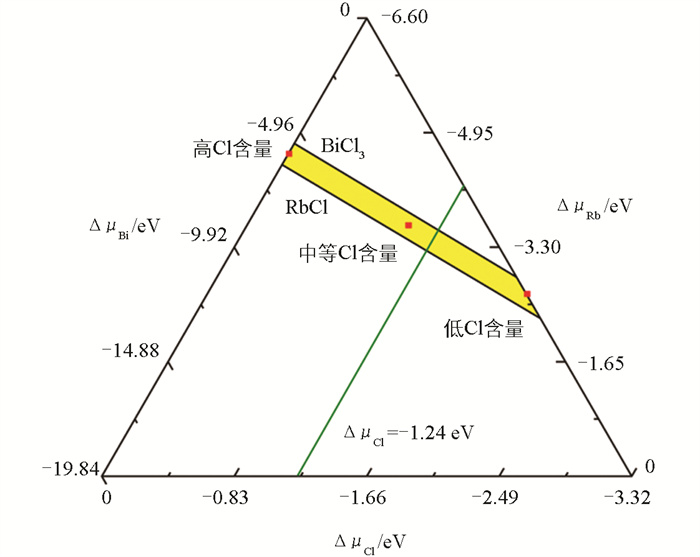
图 3是各元素在热力学平衡条件下的化学势相图。黄色区域代表可以稳定形成单一相Rb3BiCl6晶体且避免析出RbCl和BiCl3的化学势范围,ΔμRb、ΔμBi和ΔμCl的取值都为负值以确保不会形成Bi、Rb和Cl2单质。图中绿色线对应实验合成Rb3BiCl6晶体的Cl-环境(浓盐酸)下的ΔμCl,其值约为-1.24 eV。
表 1是3种不同的ΔμCl环境下计算出的部分缺陷在本征费米能级位置的最可能价态缺陷形成能。可知,随着Cl化学势的降低即合成环境中Cl含量的减少,Cl空位缺陷更容易形成,而Cl间隙缺陷更难生成。因为在低Cl含量条件下,Cl-嵌入Rb3BiCl6晶体并生成Cl间隙缺陷需要克服更高的能量,这符合化学平衡原理。经过初步筛选,形成能低的iCl-、BiRb2+比较有研究意义,其可以归因于合成环境中的Cl含量偏高(浓盐酸)且晶格内存在较大的八面体间隙。其余形成能过高的VBi3-、RbBi2-等缺陷不易形成,下文将不再进行讨论。
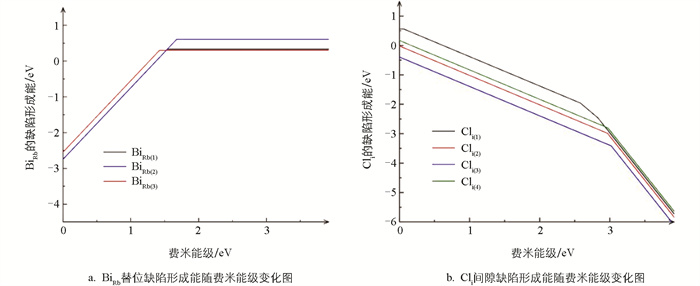
图 4分别是3个BiRb替位缺陷和4个Cli间隙缺陷位点形成能随费米能级变化图,折线斜率代表了费米能级处于该值时缺陷最可能的价态。横轴取值范围为禁带宽度,将价带顶位置设置为参考点。4个Cli间隙缺陷位点分别取自八面体空隙和层间间隙且规避了等效位点,折线的拐点处即为缺陷转变能级的位置,与导带底的间距皆大于1 eV,属于深能级缺陷。各BiRb缺陷能级较深,可能是因为高电荷Bi3+离子处于低对称性的配位环境中,6s2孤对电子高度局域化。亦有研究指出类似结构的Rb4CdCl6中CdRb缺陷也是深能级缺陷,且(+1/0)和(0/-1)转变能级距离很近,与Rb3BiCl6中只有(+2/0)转变能级的计算结果类似[13]。从整体上讲,Rb3BiCl6容易形成深能级缺陷也与该材料本身的软晶格特性和强电子-声子耦合效应相关。该材料原子间化学键较弱,点缺陷周围的原子会发生显著且局域的结构弛豫来响应缺陷,这个局域环境会强烈地“捕获”与缺陷相关的电子态,使其电子波函数被紧紧地局限在缺陷中心附近,而不是扩展到整个材料范围。这种强局域波函数往往会导致较深的缺陷转变能级[14]。同时强电子-声子耦合作用也导致缺陷不同的电荷态抛物线能量曲线发生巨大的水平偏移,电子可以直接通过一个低势垒甚至无势垒的多声子发射路径,从高能态无辐射弛豫到低能态。
值得一提的是,考虑到存在激子结合能,缺陷转变能级参考的电学带隙一般稍大于光学带隙,带隙由PBE泛函计算的结果为3.92 eV。由三维周期性体系广泛认可的PBE0杂化泛函计算的结果为5.01 eV,与实验UV-vis吸收光谱给出的光学带隙[4]估算结果3.20 eV的差距过大,因而不予采信。
图 5为考虑SOC效应下Cli缺陷和BiRb缺陷的能带图,横穿禁带的线可以近似看作是缺陷转变能级位置,纵轴能量为0的点代表本征费米能级所在位置。间隙Cl缺陷带位于本征费米能级之上且靠近价带,BiRb替位缺陷带则靠近禁带中心,两带与导带底间距离皆大于0.8 eV,验证了其最易形成深能级缺陷。一般只有浅能级缺陷束缚激子时才有可能形成高效的本征缺陷发光,而深能级缺陷对激子具有极强的“俘获”能力,一旦激子被“捕获”,其能量极有可能通过多声子发射过程以热的形式耗散,而非通过辐射跃迁释放光子[15]。这一机制从根本上截断了通过缺陷态进行辐射复合的路径,导致PL光谱中无法找到缺陷相关的发光峰。该材料易形成深能级缺陷的特性,可能与其软晶格和强电子-声子耦合有关,这进一步促进了非辐射弛豫过程[16]。以上结果表明,该材料合成过程中若生成缺陷,则基本可以确定为深能级缺陷,与实验中[4]并未观察到缺陷相关的发光结果相互验证。
-
根据激子自旋性质的不同,s-p激发态通常可分为单重态和三重态,在该晶体中最低激发态为三重态自陷态[4]。计算结果显示,自陷态总能相比自由态低约0.23 eV,属正常范围,其垂直发射能约为2.91 eV,可与实验中420 nm的发射峰较好地对应[4],激子空间局域化特征明显且主要分布在某些[BiCl6]3-周围。该自陷态的形成源于强电子-声子耦合引起的晶格弛豫,使得激发态能量以非辐射跃迁耗散的概率大幅降低[17],有利于辐射复合的发生。这一机制解释了实验中观察到的较强宽带发射现象。
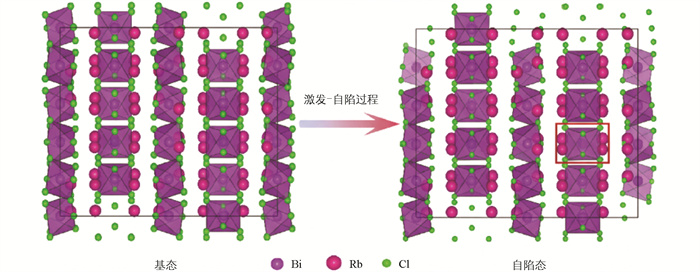
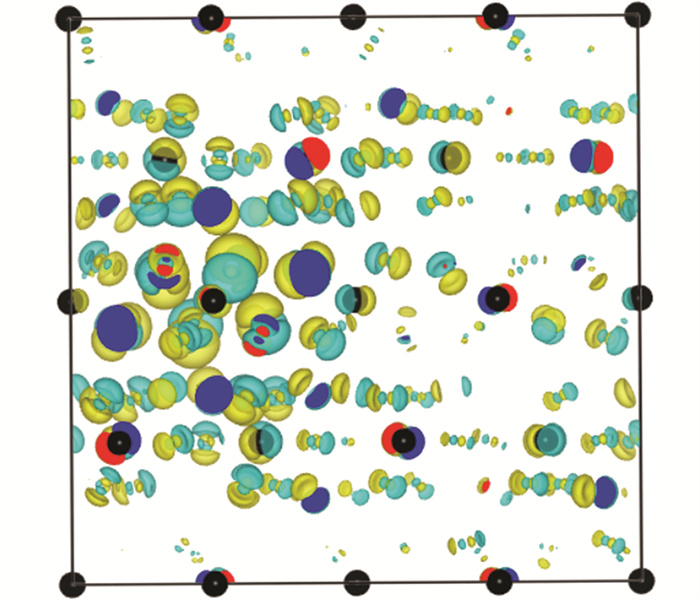
图 6为基态和自陷态结构沿某晶格方向的截面对比图。红色方框内的区域即主导自陷态形成的[BiCl6]3-八面体截面图,该八面体轻微膨胀,而其他八面体结构基本没有变化。该[BiCl6]3-八面体弛豫后整体形状与周围八面体差异也并不明显,并不影响晶格整体结构。图 7为自陷态和基态的电荷差分密度图。黑色小球为Bi原子,黄色和青色区域分别代表晶格中负电荷和正电荷分布,显示电荷的空间分布有大范围且较为局域的电子重排。由此也可以确定该计算结果中自陷态电子-声子耦合较强,有明显的空间局域性,且伴随自陷态诱导结构弛豫。这种弛豫产生于各个[BiCl6]3-八面体被Rb隔开导致的激子强局域化,图 2中其价带顶和导带底主要由Bi或Cl贡献可以佐证这一点。
如图 8所示,T1自陷态和基态的构型图相比并没有明显变化,仅表现为[BiCl6]3-八面体的微小弛豫。自陷态形成后该晶体整体仍为正交晶系,轴角变化维持在90±1°以内,晶胞整体体积相较基态增加约7‰,基本可以认为整体结构并未发生形变。考虑到该晶体整体结构较为松散,各[BiCl6]3-之间的间隙较大,部分[BiCl6]3-八面体的体积些许增大,在动力学上是合理且有利的。在Zhou等[4]的实验结果中,自陷态对应半峰宽仅约50 nm的发射峰,Stokes位移仅100 nm左右,这一现象也侧面证实该自陷态并不涉及强晶格弛豫,与轻微结构畸变这一计算结果相互吻合。从电子结构角度,由于激发而发生s-p跃迁时,外层6s2孤对电子的稳定环境被破坏,导致Bi和周围Cl的库仑作用和共价性发生显著变化,从而强烈驱动八面体结构发生弛豫,这与Zhou等[4]的fs-TA图中本征自陷态的较快弛豫时间相符。
从分子轨道[18]角度讲,基态下Bi的6s2电子实际占据的是能量较低的成键轨道,该轨道具有显著的Cl配体特征。这意味着在基态中,6s电子对Bi-Cl成键轨道有贡献,即增强了Bi与Cl之间的相互作用。同时,Bi的6p轨道与Cl的σ轨道形成反键轨道,但这些轨道在基态下没有电子占据。当体系被激发时,一个电子从6s轨道跃迁至6p轨道,导致体系的电子占据情况发生变化。激发到6p轨道的电子主要占据Bi-Cl的σ反键轨道,该轨道的电子分布主要集中在Bi原子上,且在Bi-Cl键轴上与Cl的轨道呈相位相反的重叠。该激发态的电子云呈现球对称分布,导致各方向Bi-Cl键被均匀削弱,即[BiCl6]3-中各Bi-Cl键更倾向于对称式膨胀,因为某一方向键拉长会导致其他方向键长的显著增加,从而破坏σ反键轨道的简并性[19]。因此Rb3BiCl6的自陷态对应[BiCl6]3-的对称弛豫,并没有发生质心Bi的偏移或某两个Bi-Cl键的大幅拉长。对某个单独的[BiCl6]3-,八面体键长畸变参数Δd可由以下公式确定[7]:
式中:dn为第n个Bi-Cl键长; davg为平均键长。
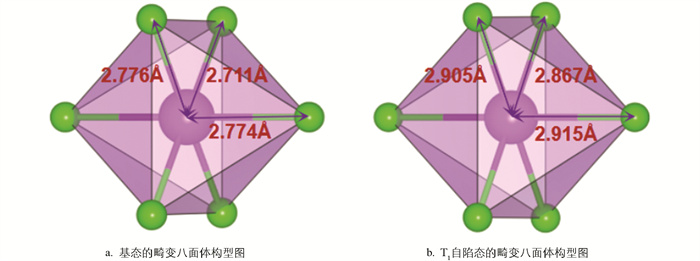
计算得到的基态、三重自由态、三重自陷态的Δd如表 2所示,表明三重自陷态的[BiCl6]3-畸变幅度比起基态和三重自由态而言略微降低。基态的[BiCl6]3-八面体呈现轻微压扁的D4h对称结构,这是由于Bi3+的6s2惰性电子对效应:6s电子对占据低能量成键轨道,使八面体沿某一轴向收缩,形成2个较短的Bi-Cl键和4个较长的Bi-Cl键。当体系被激发,一个电子从6s轨道跃迁至6p轨道,破坏了6s2惰性电子的稳定环境,导致Bi与周围Cl之间的库仑作用和共价性发生显著变化。激发后产生的激子之间存在强大的库仑引力,在对称性更高的八面体环境中,Bi的6p轨道与周围Cl的轨道相互作用更加充分,为激子波函数提供更均匀更对称的势场,使得空穴和电子的波函数在空间上能更好地重叠[20]。这一结果也说明了电子跃迁时[BiCl6]3-没有发生较大的键长改变,与Stokes位移相对较窄这一现象一致。
2.1. 能带和态密度
2.2. 本征缺陷
2.3. 自陷态发射
-
本研究通过第一性原理计算明确揭示了Rb3BiCl6的光致发光机理,定量计算了所有可能缺陷态在禁带中的能级位置,发现其缺陷能级均位于禁带深处,从理论上证实了该材料不存在缺陷辅助发光。同时,通过计算自由态与自陷态的能量差,结合[BiCl6]3-八面体畸变的原子级结构分析,明确了420 nm发光源于[BiCl6]3-单元的局域弛豫而非整体结构相变,为Bi基卤化物钙钛矿的发光调控提供了精准的理论靶点。本研究结果明确了Bi基卤化物钙钛矿发光材料中自陷态的作用,为设计高效零维发光材料提供了有价值的理论依据。