


 下载:
下载:

-
藿芪灌注液是由淫羊藿、丹参、益母草等中药组成的子宫灌注剂, 其功能为催情助孕、益气壮阳、补血养阴, 在临床中运用于奶牛持久性黄体不孕症和卵巢静止[1].已有研究表明, 该制剂工艺稳定可行[2], 无刺激性[3], 能增强动物发情调节作用, 提高雌孕激素受体基因的表达量和雌性小鼠机体雌激素水平[4].为了控制本品的质量, 便于药效学研究, 本课题组采用薄层色谱法(TLC)对藿芪灌注液中的淫羊藿、丹参、红花和益母草等进行了鉴别, 用高效液相色谱法(HPLC)对本品中主要成分淫羊藿苷和黄芪甲苷进行了含量测定.
全文HTML
-
岛津LC-20AD HPLC, SPD-20A紫外检测器:LabSolutions工作站, WondaSil C18柱(4.6 mm×250 mm, 5 μm)【岛津(中国)有限公司】; Agilent 1260 HPLC, Alltech ELSD2000ES检测器:ChemStation工作站和CSChrom Plus工作站, ZORBAX SB-C18柱(4.6 mm×150 mm, 5 μm)(安捷伦科技有限公司).
-
淫羊藿苷、黄芪甲苷(均购自中国药品生物制品检定所, 供含量测定用, 批号分别为110737-200415, 110781-200613);藿芪灌注液(中国农业科学院兰州畜牧与兽药研究所研制, 批号20120801, 20120802, 20120803, 20120804, 20120805, 20120901, 20120902, 20120903, 20120904, 20120905);乙腈、甲醇均为色谱纯(购自天津市四友精细化学品有限公司); 正丁醇、乙酸乙酯、丁酮、甲酸、乙醚、盐酸、硫氰酸铬铵和丙酮等试剂均为分析纯, 水为超纯水.
1.1. 仪器
1.2. 试剂
-
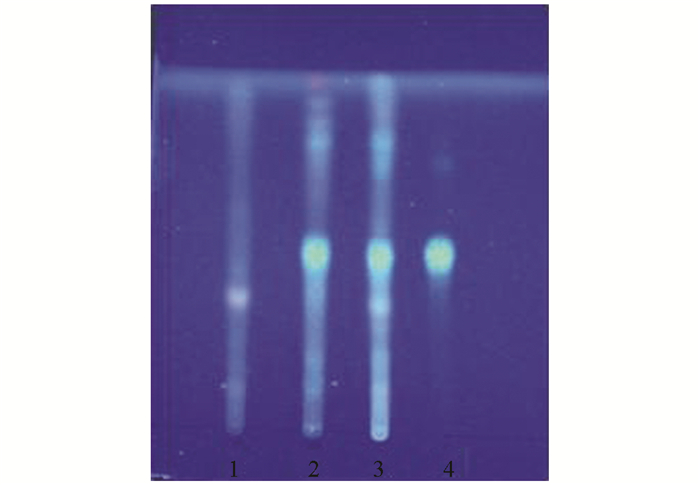
量取本品10 mL, 蒸干; 残渣加70%乙醇30 mL, 用1 h进行超声, 过滤, 滤液蒸干; 用30 mL水溶解残渣, 振摇萃取3次(水饱和的正丁醇溶液作为溶剂), 每次20 mL, 合并正丁醇萃取液; 洗涤3次(1%氢氧化钠溶液), 每次20 mL, 弃去洗液; 再洗涤2次(正丁醇饱和的水溶液), 每次15 mL, 蒸干正丁醇洗涤液, 用甲醇1 mL使残渣溶解, 作为供试液; 另取淫羊藿对照药材1 g, 加30 mL 70%乙醇, 用1 h进行超声, 过滤, 同样方法制成对照药材溶液; 再取对照品淫羊藿苷, 加甲醇制成0.3 mg/mL的溶液, 作为对照品溶液.按照0502薄层色谱法试验, 吸取上述3种溶液各2 μL, 分别点于同一硅胶G薄层板上, 展开剂为乙酸乙酯-丁酮-甲酸-水(10:1:1:1), 展开, 取出, 晾干, 喷显色剂(5%三氯化铝乙醇溶液), 加热3分钟(105 ℃), 置紫外光灯(365 nm)下检视.供试品、对照药材和对照品色谱在相应的位置上, 具有同种颜色的荧光斑点(图 1).
-
量取本品20 mL, 振摇提取1次(20 mL水饱和的正丁醇溶液作为溶剂), 有机层弃去, 用稀盐酸调水层pH值至2.0;水层振摇提取2次(乙醚溶液), 每次20 mL, 合并有机层, 挥干; 用2 mL甲醇使残渣溶解, 作为供试液.取对照药材丹参1 g, 加水20 mL并加热回流1 h, 滤过, 滤液加水至20 mL, 用同样方法制成对照液.按照0502薄层色谱法试验, 吸取供试液1 μL、对照液2 μL, 分别点于同一硅胶G薄层板上, 展开剂为甲苯-三氯甲烷-乙酸乙酯-甲醇-甲酸(2:3:4:0.5:2), 展开, 取出, 晾干, 喷显色剂(含5%香草醛的10%硫酸乙醇溶液), 105 ℃加热显色至斑点清晰.供试品与对照药材色谱在相应的位置上, 显同种颜色的斑点(图 2).
-
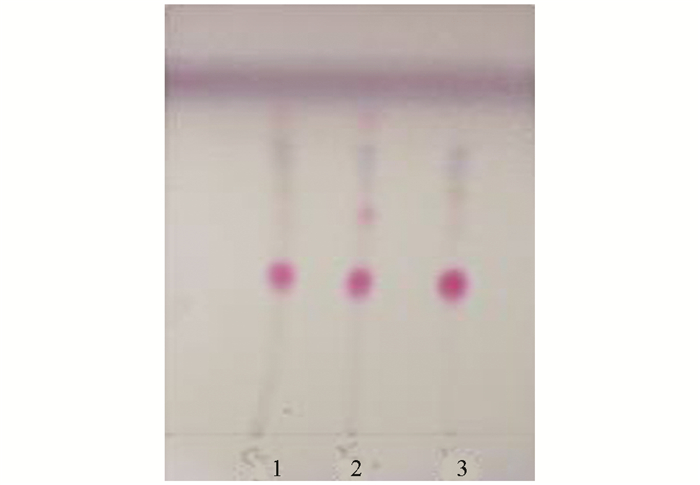
量取本品20 mL, 振摇提取3次(水饱和正丁醇溶液作为溶剂), 每次20 mL, 合并正丁醇液, 蒸干; 加甲醇5 mL使残渣溶解, 将溶解液加入中性氧化铝柱(100~200目, 4 g, 内径1.5 cm), 用20 mL甲醇进行洗脱, 收集洗脱液, 蒸干; 加甲醇4 mL使残渣溶解, 作为供试液.取不含红花药材的本品阴性对照液, 按供试液同样方法制得阴性对照品溶液.另取对照药材红花1 g, 加水200 mL, 加热回流1 h, 过滤, 滤液浓缩至20 mL, 同样方法制成对照液.按照0502薄层色谱法试验, 吸取供试液5 μL, 对照液10 μL, 分别点于同一硅胶G薄层板上, 展开剂为环己烷-乙酸乙酯(5:3), 展开, 取出, 晾干, 喷显色剂(含5%香草醛的10%硫酸乙醇溶液), 105 ℃加热至斑点显色清晰.供试品与对照药材色谱在相应的位置上, 显同种颜色的斑点(图 3).
-
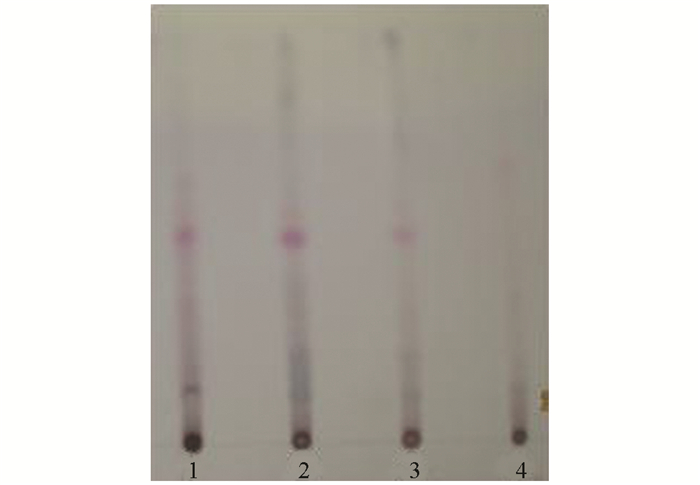
量取本品10 mL, 加浓盐酸调pH值至2.0, 离心除去沉淀, 少量水洗涤沉淀, 洗液合并入上清液; 上清液中加入新鲜配制的2%硫氰酸铬铵溶液10 mL, 冰浴中放置l h后离心10 min, 弃去上清液, 沉淀用少量冰水洗涤后, 加入2 mL丙酮溶解; 丙酮液中滴加0.5%硫酸银溶液6 mL, 离心10 min, 倾出上清液, 沉淀用少量丙酮洗涤; 合并洗液与上清液并浓缩至约l mL, 滴加1%氯化钡溶液2 mL, 离心10 min, 倾出上清液, 沉淀用50%乙醇洗涤; 合并上清液与洗液并蒸干, 残渣加甲醇2 mL溶解, 作为供试液.另取盐酸水苏碱对照品, 加无水乙醇制成1 mL/mg的溶液, 作为对照液.取不含益母草药材的本品阴性对照液, 按供试液同样制得阴性对照品溶液.按照0502薄层色谱法试验, 吸取上述3种溶液各5 μL, 分别点于同一硅胶G薄层板上, 展开剂为丙酮-无水乙醇-盐酸(10:6:1), 展开, 取出, 晾干, 105 ℃加热15 min, 放冷, 喷显色剂(稀碘化铋钾试液-三氯化铁试液(10:1))至斑点显色清晰.供试品与对照药材色谱在相应的位置上, 显同种颜色的斑点(图 4).
-
(1) 色谱条件
色谱柱:WondaSil C18柱(4.6 mm×250 mm, 5 μm), 流动相:乙腈-水(28:72), 流速:1.0 mL/min, 检测波长:270 nm, 柱温:40 ℃, 进样量:10 μL, 以淫羊藿苷计理论塔板数不低于1 500.
(2) 样品溶液的制备
对照品溶液:精密称取对照品淫羊藿苷11.68 mg, 置50 mL容量瓶中, 用色谱甲醇定容至刻度, 摇匀, 制得233.6 μg/mL的对照品储备溶液.精密移取储备液5 mL于10 mL容量瓶中, 用甲醇定容至刻度, 制得116.8 μg/mL的对照液①, 吸取对照液①1 mL于10 mL容量瓶中, 用甲醇定容至刻度, 得质量浓度为11.68 μg/mL的对照液②.
供试品溶液:精密量取本品1 mL于25 mL容量瓶中, 加水定容至刻度, 摇匀, 滤过, 取续滤液, 即得供试液.
阴性对照溶液:取按处方比例并以相同工艺制备的缺淫羊藿药材的阴性对照供试品, 按供试品溶液制备法制得阴性对照溶液.
(3) 专属性试验
取2.2.1(2)项下对照品、供试品和阴性对照溶液, 过0.22 μ有机微孔滤头于1 mL进样瓶中, 密封, 按照HPLC测定法并按2.2.1(1)项色谱条件自动进样测定, 每个样品进样2次, 记录色谱图.在与淫羊藿苷对照品溶液色谱峰对应的位置上, 供试品与其对照品保留时间一致, 且与其他组分分离度大于1.5;阴性对照溶液在对照品与供试液相应的保留时间内未检出色谱峰, 表明样品中其他成分不干扰淫羊藿苷的测定.
(4) 线性关系考察
吸取对照液①1 mL分别于50 mL, 25 mL, 10 mL, 5 mL, 2 mL容量瓶中, 用甲醇稀释至刻度, 制成质量浓度分别为2.336 μg/mL, 4.672 μg/mL, 11.68 μg/mL, 23.36 μg/mL, 58.4 μg/mL的对照品溶液.按上述色谱条件, 对照品溶液依次进样10 μL, 记录峰面积.纵坐标为峰面积, 横坐标为对照品质量浓度, 绘制标准曲线, 得y=23 054.98x+1 386.77, r=0.999 9, 表明淫羊藿苷对照品在2.336~58.4 μg/mL范围内与峰面积积分值呈良好的线性关系.
(5) 定量限与检测限确定
精取对照品溶液, 用甲醇逐级稀释定容, 按上述色谱条件进样10 μL, 记录色谱图.信噪比为10:1时测得淫羊藿苷定量限为2.026 ng/mL, 信噪比为3:1时测得检测限为0.506 5 ng/mL.
(6) 重复性试验
按2.2.1(2)项下供试品液制备法制备同一批次(批号:20120802)的供试品溶液6份, 按上述色谱条件进样, 记录峰面积并计算含量、 RSD 值.得淫羊藿苷的含量均值为0.232 mg/mL, RSD =0.521%, 表明本方法重复性良好.
(7) 精密度试验
(a) 日内精密度试验
取对照液②, 按上述色谱条件重复进样6次, 得淫羊藿苷平均峰面积为277 594, RSD =0.287%, 结果表明日内精密度良好.
(b) 日间精密度试验
取对照液②10 μL, 按上述色谱条件连续3 d进样, 每天重复进样2次.得淫羊藿苷平均峰面积为275 099, RSD =0.777%, 结果表明日间精密度良好.
(8) 稳定性试验
精密量取供试液, 在室温放置0, 1, 2, 3, 6, 9, 12 h后分次按上述色谱条件测定淫羊藿苷含量, 计算 RSD .计算结果7次进样的淫羊藿苷含量平均值为0.236 mg/mL, RSD =0.969%(n=7), 表明供试液中淫羊藿苷的含量在12 h内保持稳定.
(9) 加样回收率试验
精密吸取同批次(批号:20120802)6份已知含量的藿芪灌注液样品1.0 mL, 各精密加入0.280 6 mg/mL淫羊藿苷对照品溶液1.0 mL于25 mL容量瓶中, 加水稀释至刻度, 摇匀, 滤过, 取续滤液, 即得加样回收供试液.按上述色谱条件测定淫羊藿苷含量并计算回收率和 RSD .其平均回收率为100.2%, RSD =0.45%, 表明回收率良好(表 1).
(10)样品含量测定
分别取10批藿芪灌注液(批号20120801, 20120802, 20120803, 20120804, 20120805, 20120901, 20120902, 20120903, 20120904, 20120905), 按2.2.1(2)项供试液法每批次各平行制备2份供试液, 按色谱条件自动进样测定, 记录色谱图和峰面积, 并计算淫羊藿苷含量和 RSD 值.得淫羊藿苷的的平均含量为0.242 mg/mL, RSD =7.49%.考虑到大规模生产中的各种损失和测量误差, 取其平均值的80%作为其含量限度, 即得0.193 6 mg/mL, 故暂定本品含淫羊藿按淫羊藿苷计不少于0.20 mg/mL.
-
(1) 色谱条件
色谱柱:ZORBAX SB-C18柱(4.6 mm×150 mm, 5 μm), 漂移管温度为90 ℃, 载气流速为2.5 L/min, 流动相:甲醇-水(65:35), 流速:1 mL/min, 柱温:40 ℃, 检测用蒸发光散射检测器.理论板数按黄芪甲苷峰计算应不低于3 000.
(2) 样品溶液的制备
对照品溶液:精密称取对照品黄芪甲苷11.95 mg于10 mL容量瓶中, 加甲醇溶解并定容至刻度, 摇匀, 制得1.195 mg/mL的对照品储备液, 精密量取对照品储备液1 mL于5 mL容量瓶中, 加甲醇定容至刻度, 摇匀, 制成质量浓度为0.239 mg/mL的对照液.
供试品溶液:精密量取本品25 mL, 振摇提取4次(水饱和的正丁醇溶液作为溶剂), 每次40 mL, 弃去水层, 合并正丁醇提取液; 洗涤2次(氨试液), 每次25 mL, 弃去氨液, 取正丁醇液, 挥干溶剂, 用甲醇使残渣溶解并移至5 mL容量瓶中, 加甲醇定容至刻度, 摇匀, 滤过, 取续滤液, 即得.
阴性对照溶液:取按处方比例并以相同工艺制备的缺黄芪的阴性对照供试品, 按供试品溶液制备法制得阴性对照液.
(3) 专属性试验
取2.2.2(2)项下对照品、供试品和阴性对照溶液, 过0.22 μ有机微孔滤头于1 mL进样瓶中, 密封, 按照HPLC测定法并按2.2.2(1)项色谱条件自动进样测定, 每个样品进样2次, 记录色谱图.在与黄芪甲苷对照品溶液色谱峰对应的位置上, 供试品与其对照品保留时间一致, 且与其他组分分离度大于1.5;阴性对照溶液在对照品与供试液相应的保留时间内未检出色谱峰, 表明样品中其他成分不干扰黄芪甲苷的测定.
(4) 线性关系考察
精密量取对照品储备溶液1 mL, 1 mL, 1 mL, 2 mL, 分别于25 mL, 10 mL, 5 mL, 5 mL容量瓶中加甲醇稀释至刻度, 摇匀, 滤过, 制成质量浓度分别为0.0478 mg/mL, 0.1195 mg/mL, 0.239 mg/mL, 0.478 mg/mL的对照品溶液.按上述色谱条件, 对照品溶液依次进样10 μL, 测定峰面积.纵坐标用峰面积表示, 横坐标用对照品质量浓度表示, 绘制标准曲线.得y= 1.470 039x+2.340 902, r=0.999 6, 结果样品质量浓度在0.047 8~0.478 mg/mL范围内, 峰面积线性关系良好.
(5) 定量限与检测限确定
精取对照品溶液, 用甲醇逐级稀释, 按上述色谱条件进样10 μL, 记录色图谱.信噪比为10:1时测得黄芪甲苷定量限为442 ng/mL, 信噪比为3:1时测得检测限为119.5 ng/mL.
(6) 重复性试验
按2.2.2(2)项下供试品溶液制备法制备6份同批次样品(批号:20120802), 按上述色谱条件进样, 记录峰面积, 并计算含量与 RSD 值.测定得黄芪甲苷平均含量为84.31 μg/mL, RSD =1.97%, 表明该方法重复性良好.
(7) 精密度试验
(a) 日内精密度试验
取对照液, 按上述色谱条件重复进样6次.结果供试液峰面积均值为516 892.3, RSD =0.638%(n=6), 表明仪器日内精密度良好.
(b) 日间精密度试验
取黄芪甲苷对照液和供试液, 按上述色谱条件连续3 d进样, 每天重复进样2次, 记录峰面积并计算 RSD .计算结果峰面积均值分别为512 752.3, 547 107.5, RSD 分别为1.18%和1.80%(n=6), 表明仪器精密度良好.
(8) 稳定性试验
按2.2.2项下制备一份供试液(批号:20120802), 将供试液分别于室温放置0, 1, 2, 3, 5, 8 h, 精密吸取供试品溶液按上述色谱条件进样, 测定黄芪甲苷含量并计算 RSD .结果6次进样的黄芪甲苷含量平均为56.52 μg/mL, RSD =0.926%(n=6), 试验表明供试品溶液中黄芪甲苷的含量在8 h内保持稳定.
(9) 加样回收率试验
加样回收对照品液的制备:精密称定黄芪甲苷对照品18.38 mg于10 mL容量瓶中, 加甲醇定容至刻度, 制成1.838 mg/mL黄芪甲苷对照品储备液.
精密量取藿芪灌注液25 mL, 1.838 mg/mL黄芪甲苷对照品储备液1 mL, 混匀, 按2.2.2项供试液制备法制备同批次6份(批号:20120802)供试液.按上述色谱条件进样, 测定黄芪甲苷含量并计算回收率和 RSD .黄芪甲苷平均回收率为99.26%, RSD =1.46%, 表明其回收率良好(表 2).
(10)样品含量测定
分别取10批藿芪灌注液样品(批号20120801, 20120802, 20120803, 20120804, 20120805, 20120901, 20120902, 20120903, 20120904, 20120905), 按2.2.2(2)项下供试品溶液的制备方法制备样品, 按以上色谱条件测定含量.结果10批样品中黄芪甲苷的的平均含量为64.653 μg/mL, RSD =8.68%.考虑到大规模生产中的各种损失, 取其平均值的80%作为其含量限度, 即得45.55 μg/mL, 故暂定本品黄芪按黄芪甲苷计不少于45.0 μg/mL.
2.1. TLC鉴别
2.1.1. 淫羊藿
2.1.2. 丹参
2.1.3. 红花
2.1.4. 益母草
2.2. 含量测定
2.2.1. 淫羊藿苷
2.2.2. 黄芪甲苷
-
淫羊藿和黄芪被广泛应用于多种制剂之中, 在本制剂中也是作为君药发挥其药效药理作用.淫羊藿苷与黄芪甲苷分别为淫羊藿和黄芪的主要成分[5], 都发挥主要药理活性作用, 同时中国药典对其含量标准也有规定, 故对其进行方法学研究, 对完善本品的质量标准具有十分重要的作用, 也进一步控制了本品的内在质量, 对临床药效的发挥具有更重要的意义.
-
淫羊藿与黄芪作为传统中医保健用药, 定量检测方法已比较成熟, 众多报道采用HPLC对淫羊藿苷和黄芪甲苷的检测方法基本一致[6-10], 但在此试验上, 本课题组用新方法进行研究并得到了良好的结果.
本品参考中国兽药典淫羊藿苷含量测定方法进行研究, 考察了柱温、流速、流动相比例及波长的选择.改变柱温, 供试品溶液中淫羊藿苷含量的 RSD 为8.18%, 说明柱温对淫羊藿苷含量测定有一定的影响; 在柱温40 ℃条件下, 改变流速, 供试品溶液中淫羊藿苷含量的 RSD 为1.04%, 说明在柱温40 ℃条件下, 改变流速对样品中淫羊藿苷的测定无影响.因此, 试验证明以流动相乙腈-水(28:72)、检测波长270 nm、柱温40 ℃、流速1.0 mL/min的色谱条件对淫羊藿苷的测定具有较强的专属性, 能达到基线平衡、分离度较高、具有对称的峰形、阴性对照无干扰且缩短了分析时间, 便于作为指纹图谱鉴别[11].
黄芪甲苷作为黄芪的主要成分之一, 仅在200 nm左右有弱的紫外末端吸收, 因而用紫外检测器(VWD)操作比较困难, 且噪音对结果影响较大, 灵敏度也较低, 故HPLC-VWD法一直未能广泛用于黄芪甲苷的测定[12-13].蒸发光散射检测(ELSD)由被分析物质颗粒的数量和大小决定光散射检测器的响应大小, 而不受流动相溶剂的干扰, 此种检测器可用于非挥发性成分的检测[14].本研究采用HPLC -ELSD法对黄芪甲苷进行含量测定, 考察了流动相比例、流速及温度的变化, 两个供试液中黄芪甲苷含量的 RSD 分别为1.77%, 1.40%, 说明测定条件小范围变动, 对样品中黄芪甲苷含量的测定无影响.综合考虑, 最终确定色谱条件为漂移管温度90 ℃, 载气流速2.5 L/min, 柱温:40 ℃, 流动相:甲醇-水(65:35), 流速:1 mL/min, 其所测得结果灵敏度高、分离度良好、重现性好, 是一种良好的黄芪甲苷测定方法[15-16].
-
中药化学成分复杂多样, 加之本制剂是由多种中药混煎而得, 因此供试液前处理是目前中药制剂主要组分含量测定必不可缺的步骤.从本试验研究可以看出, 供试液的前处理可能显著影响其成分含量测定的结果及薄层色谱的鉴别.因此, 在研究中药制剂质量标准时, 不仅要考虑待测指标的含量, 还应考察前处理方法对测定结果的影响[17-18].
含淫羊藿制剂的前处理方法一般使用超声、回流及溶剂直接定容[19].由于本制剂为液体剂型, 不存在被检成分能否完全溶出的问题, 因此在供试液制备时首选操作简便的直接溶剂稀释法, 使用了甲醇、乙腈及超纯水定容稀释[20].研究表明, 取1 mL样品进行适当稀释后, 在线性范围内可对该制剂中的淫羊藿苷进行准确检测, 且用超纯水与甲醇、乙腈相比较, 其淫羊藿苷含量无明显差距.故建立淫羊藿苷的含量测定方法, 可直接用超纯水定容, 其方法具有高效省时、成本低廉、结果准确、重现性好的优点.在TLC鉴别时同样采用了原液点板的方法, 但置紫外灯下检查干扰性大, 均无结果, 故采取了有机溶剂萃取法, 且在喷显色剂后再置紫外灯下观察, 其结果均无干扰, 重复性好, 图谱分离度高.
黄芪有效成分的提取多采用高速离心、超声波微波、超临界CO2萃取、高速逆流等技术[21], 但在HPLC法中测定黄芪甲苷含量时, 使用这些方法使其提取的成分太多, 干扰性太大, 故选取萃取的方法处理[22].使用多种萃取剂后, 最后选择了水饱和的正丁醇, 较其他萃取剂相比其出峰效果良好, 含量稳定.在其他相同条件下进行了提取次数的优选, 分别振摇提取3, 4, 5次, 试验表明用供试品提取3次与4次、5次有较大差异, 而4次、5次时黄芪甲苷含量无明显差异, 故选择用水饱和的正丁醇溶液振摇提取4次.所用氨试液, 主要为了除去制剂中的极性成分, 减少杂质干扰, 进而增加系统分离能力[23].将提取液蒸干再用甲醇溶解进样, 对系统ELSD分解度较好, 能更好地分离黄芪甲苷, 所得到的黄芪甲苷含量也更加可观, 故使用该方法能够有效控制本制剂中黄芪甲苷的含量, 对制定其质量标准提供了一定的指导作用[24].
作为复方中药制剂的鉴别, 应根据其药材化学成分, 寻找简便快速并具有专属性较强的方法, 才能起到有效控制质量的目的[25].鉴别本品处方中丹参时, 经过与之前研究比较得出样品与药材采用正丁醇除杂, 乙醚提取后, 斑点更为明显, 更适合丹参有效成分的提取, 但斑点分散, 故进行展开剂比例调整, 其后发现供试品色谱中与药材色谱相对应的斑点会随着展开剂的变化同步移动, 药材色谱中比供试品色谱上方多一个斑点, 疑似在样品的制备过程中采用了醇沉和灭菌工艺致使有效成分被破坏, 故改用样品的制备工艺处理丹参药材, 以确定供试品溶液与药材溶液斑点是否为同一成分, 并比较硅胶GF254薄层板与硅胶G薄层板展开效果.结果表明醇沉、灭菌对丹参药材成分影响较大, 供试品色谱中只能与药材色谱有一个相应斑点对应, 原因是经醇沉、灭菌后所致, 故最终选定与药材色谱下方斑点相对应即可达到鉴别效果.
鉴别本品处方中红花时, 比较不同提取方法制备的药材溶液, 结果表明药材溶液需通过中性氧化铝柱或D101型大孔吸附树脂柱方可出现粉红色斑点[26], 但中性氧化铝柱的操作方法比大孔树脂柱简单、省时, 显色效果也更清晰, 故选择中性氧化铝柱方法为本试验最终方法.
鉴别本品处方中益母草时, 参考中国药典、中国兽药典方法处理供试液, 且尝试用了732型强酸性阳离子交换树脂柱[27-28], 其供试品溶液、对照药材溶液在与对照品溶液相同位置上出现相应斑点, 但是背景有点干扰, 其色谱图分离度不高, 有拖尾现象, 经查阅文献及多次试验筛选最终确定将其药液调整pH值后用硫氰酸铬铵试液进行处理, 再用丙酮、硫酸银及氯化钡溶液依次进行洗涤溶解等, 其显色效果更加清晰, 分离度高, 阴性对照无干扰[29-30].
3.1. 待测含量组分选择
3.2. 色谱条件选择
3.3. 优化供试液前处理法
-
通过以上方法研究的验证结果表明:所建立的特征图谱专属性强、准确可行、简便, 可快速用于定性鉴别及定量检测, 可广泛用于该制剂的质量控制方法之中.