


 下载:
下载:







-
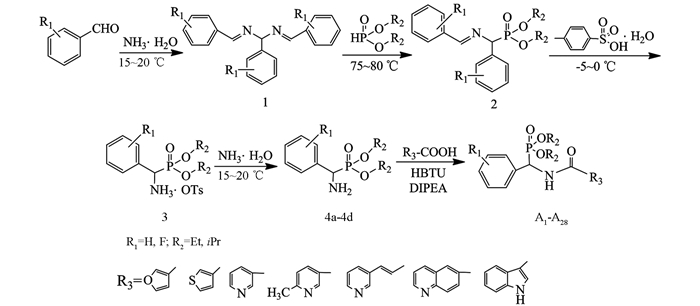
杂环化合物因其具有种类繁多、结构多样和生物活性广泛等特点,不仅普遍存在于天然药物分子中,同时在人工合成化学药物中也被广泛应用[1-2].据统计,目前临床使用的90%以上的药物为杂环化合物,因此,从大量的杂环化合物中寻找具有高疗效、低毒性、容易获得的候选物仍然是当前新药研发的热点之一[3-4].另一方面,含磷化合物广泛的生物活性及多变的结构类型一直受到人们的关注,许多天然产物药物分子以及合成药物都含有磷原子.近年来本课题组先后设计合成一系列含膦酸酯、膦酰基、硫脲膦酸酯类化合物,发现不同系列化合物表现出抗病毒[5-8]、抗肿瘤[9-12]、杀菌[13-14]等生物活性.其中α-氨基膦酸酯,作为天然氨基酸的含磷类似物,已成为有机磷化学发展的重要组成部分,在药物合成中扮演着重要的角色,是一些药物合成的关键中间体.人们通过对其合成方法[15-17]与生物活性[18-21]的研究,发现其对肾素合成酶、HIV酶、FPT酶、EPSP合成酶及高血压蛋白原酶等具有抑制作用[15, 17-18],从而表现出多种重要的生理活性,广泛应用于医药和农药领域.由此,为了拓宽化合物生物活性并寻找到高活性先导物,基于前期的研究工作[7-13],我们将不同杂环引入氨基膦酸酯中,并优化反应条件,合成了一系列新型杂环膦酸酯衍生物28个,其结构均经IR,(2H,13C,31P,19F)NMR和HRMS(ESI)等确证与表征,合成路线见图 1.生物活性测试结果表明,部分化合物具有抗大肠杆菌、绿脓杆菌或金黄色葡萄球菌等活性;同时,发现化合物A16,A24,A28对肿瘤细胞的抑制活性较好,与对照药剂辛二酰苯胺异羟肟酸(SAHA)接近.在此基础上研究其构效关系,对进一步结构改造与优化具有十分重要的意义.
全文HTML
-
B11-1型磁力搅拌器,上海司乐仪器有限公司;N-110型旋转蒸发仪,上海爱朗仪器有限公司;X-4型显微熔点测定仪,上海易测仪器设备有限公司;Shimadzu IR Prestige-21型傅立叶红外光谱测定,日本岛津公司(KBr压片法);JEOLECX400型400 MHz核磁共振仪,日本电子株式会社(TMS为内标,CDCl3或DMSO-d6为溶剂);UPLC-QTOF Xevo G2-XS电喷雾四级杆串联飞行时间质谱仪,美国沃特世公司.所用试剂均为市售AR或CP级,使用前经常规方法处理.
-
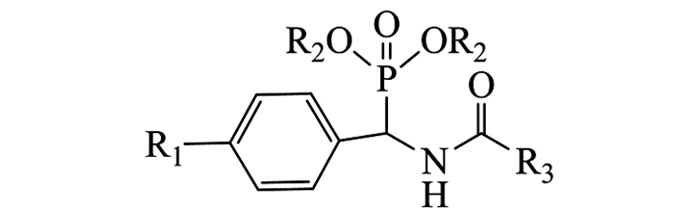
在50 mL三口瓶中加入0.5 mmol杂环甲酸化合物(固体),用5 mL无水干燥THF溶解后加O-苯并三氮唑-四甲基脲六氟磷酸酯(HBTU 0.5 mmol,1eq),混合均匀后再加催化剂N-N-二异丙基乙胺(DIPEA 1 mmol,2eq)及中间体4(0.5 mmol).在油浴加热保持微沸腾(66 ℃左右)中回流并搅拌反应,底物由浑浊变为澄清,以TLC跟踪反应5~6 h后停止反应.粗产物经薄层色谱(CH2Cl2/C2H5OH/NH3·H2O为10/1/0.1)分离纯化得到目标产物A1-A28,合成路线见图 1,结构通式见图 2,取代基、产率与熔点结果见表 1.
-
O,O'-二异丙基-α-(4-氟苯基)-α-(3-呋喃甲酰胺基)甲膦酸酯(A4):2H NMR(400 MHz,CDCl3)δ:8.01(s,1H,NH),7.57~7.35(m,4H,ArH),6.98(t,J=8.2Hz,2H,ArH),6.73(s,1H,ArH),5.61(dd,J=21.9,9.4Hz,1H,NCH-P),4.72~4.46(m,2H,2CH),1.31(d,J=6.1Hz,3H,CH3),1.27~1.22(m,6H,2CH3),0.92(d,J=6.1Hz,3H,CH3);13C NMR(101 MHz,CDCl3)δ:162.1,145.4,143.7,131.6,130.4,130.3,130.3,122.0,115.6,115.4,108.7,72.6,72.5,49.2,24.3,24.2,23.9,23.2;19F NMR(376 MHz,CDCl3)δ:-113.9 mg/kg;31P NMR(162 MHz,CDCl3)δ:20.3 mg/kg;HRMS(ESI) C18H23FNO5P[M+H]+理论值:384.137 9,实际值:384.137 6.
O,O'-二异丙基-α-(4-氟苯基)-α-(6-甲基-3-吡啶甲酰胺基)甲膦酸酯(A16):2H NMR(400 MHz,CDCl3)δ:8.94(s,1H,ArH),8.33~8.18(m,1H,ArH),7.99(dd,J=8.1,1.5Hz,1H,NH),7.66~7.44(m,2H,ArH),7.10(d,J=8.0Hz,1H,ArH),6.95(t,J=8.1Hz,2H,ArH),5.64(dd,J=21.8,9.2Hz,1H,NCH-P),4.76~4.38(m,2H,2CH),2.53(d,J=1.2Hz,3H,CH3),1.27(dd,J=6.1,1.3Hz,3H,CH3),1.24~0.86(m,9H,3CH3);13C NM(101 MHz,CDCl3)δ:165.6,161.9,148.3,135.7,131.4,130.5,130.4,130.3,126.9,122.8,115.6,115.4,72.6,72.6,49.7,24.6,24.3.19F NMR(376 MHz,CDCl3)δ:-113.8 mg/kg;31P NMR(162 MHz,CDCl3)δ:19.9 mg/kg;HRMS(ESI) C20H26FN2O4P[M+H]+理论值:409.168 9,实际值:409.169 2.
O,O'-二异丙基-α-苯基-α-(6-喹啉甲酰胺基)甲膦酸酯(A22):2H NMR(400 MHz,CDCl3)δ:8.96(dd,J=4.2,1.7Hz,1H,ArH),8.34(d,J=1.6Hz,1H,ArH),8.19~8.15(m,1H ArH),8.13~8.10(m,2H ArH,NH),7.87(dd,J=9.1,4.5Hz,1H,ArH),7.62~7.57(m,2H,ArH),7.44(dd,J=8.3,4.2Hz,1H,ArH),7.36~7.27(m,3H,ArH),5.77~5.67(m,1H,NCH-P),4.782~4.34(m,2H,2CH),1.31(d,J=6.2Hz,3H,CH3),1.25(d,J=6.2Hz,3H,CH3),1.19(d,J=6.2Hz,3H,CH3),0.83(d,J=6.2Hz,3H,CH3);13C NMR(101 MHz,CDCl3)δ:166.4,152.1,149.4,137.3,135.6,132.0,129.9,128.7,128.7,128.7,128.6,128.3,128.3,128.1,127.7,127.6,122.0,72.7,50.7,24.3;31P NMR(162 MHz,CDCl3)δ:20.4 mg/kg;HRMS(ESI) C23H27N2O4P[M+H]+理论值:427.177 8,实际值:427.178 7.
O,O'-二异丙基-α-(4-氟苯基)-α-(6-喹啉甲酰胺基)甲膦酸酯(A24):2H NMR(400 MHz,CDCl3)δ:8.97~8.93(m,1H,ArH),8.34(s,1H,ArH),8.12(m,4H,ArH,NH),7.63~7.43(m,3H,ArH),7.00(dd,J=12.1,4.8Hz,2H,ArH),5.77~5.66(m,1H,NCH-P),4.76~4.37(m,2H,2CH),1.28(dd,J=6.2,1.6Hz,3H,CH3),1.21(m,6H,2CH3),0.88(dd,J=6.2,1.3Hz,3H,CH3);13C NMR(101 MHz,CDCl3)δ:166.6,152.1,149.3,137.3,131.9,131.6,131.5,130.5,130.4,129.8,128.3,127.6,122.0,115.7,115.5,72.7,72.6,49.9,24.3;19F NMR(376 MHz,CDCl3)δ:-113.6 mg/kg;31P NMR(162 MHz,CDCl3)δ:20.0 mg/kg;HRMS(ESI) C23H26FN2O4P[M+H]+理论值:445.169 8,实际值:445.169 2.
O,O'-二乙基-α-(4-氟苯基)-α-(3-吲哚甲酰胺基)甲膦酸酯(A27):2H NMR(400 MHz,CDCl3)δ:9.60(s,1H,ArH),8.04(d,J=7.4Hz,1H,NH),7.78(s,1H,ArH),7.50(d,J=6.7Hz,2H,ArH),7.34 (d,J=7.6Hz,1H,ArH),7.28~7.09(m,3H,ArH),7.00(t,J=8.0Hz,2H,ArH),5.77(dd,J=21.2,9.0Hz,1H,NCH-P),4.20~3.75(m,4H,2CH2),1.28~1.13 (m,6H,2CH3);13C NMR(101 MHz,CDCl3)δ:164.8,161.3,136.5,131.4,130.0,129.8,128.9,124.9,123.1,122.0,115.9,115.6,112.3,111.0,63.8,48.6,16.5,16.3;19F NMR(376 MHz,CDCl3)δ:-113.7 mg/kg;31P NMR(162 MHz,CDCl3)δ:23.0 mg/kg;HRMS(ESI) C20H22FN2O4P[M+H]+理论值:405.137 5,实际值:405.137 9.
O,O'-二异丙基-α-(4-氟苯基)-α-(3-吲哚甲酰胺基)甲膦酸酯(A28):2HNMR(400 MHz,DMSO)δ:9.56(s,1H,ArH),8.03(d,J=7.0Hz,1H,NH),7.76(s,1H,ArH),7.50(d,J=6.8Hz,2H,ArH),7.37(d,J=7.5Hz,1H,ArH),7.30~7.17(m,3H,ArH),7.00(t,J=7.9Hz,3H,ArH),5.67(dd,J=21.3,8.5Hz,1H,NCH-P),4.73~4.51(m,2H,2CH),1.38~0.91(m,12H,4CH3);13C NMR(101 MHz,DMSO)δ:163.2,161.3,136.5,132.0,130.1,128.6,124.8,123.1,120.0,115.7,112.1,111.6,72.6,72.2,49.3,24.3,23.9,23.3;19F NMR(376 MHz,DMSO)δ:-114.1 mg/kg;31P NMR (162 MHz,DMSO)δ:20.9 mg/kg;HRMS(ESI) C22H26FN2O4P[M+H]+理论值:433.169 1,实际值:433.169 2.
1.1. 仪器与试剂
1.2. 化合物的合成
1.2.1. 中间体4的合成
1.2.2. 目标物A的合成
1.2.3. 部分目标化合物A的核磁、高分辨质谱数据
-
供试菌及来源:普通标准菌株大肠杆菌E.coli(ATCC25922),铜绿假单胞杆菌P.aeruginosa (ATCC9027),金黄色葡萄球菌S.aureus(ATCC6538)来源于广东省微生物保藏中心;多重耐药性细菌:N-E.coli-耐药性大肠杆菌(20151027074),N-P.aeruginosa-耐药性铜绿假单胞杆菌(20151025026),MRSA-耐甲氧西林金黄色葡萄球菌(20151027077),均于贵州医科大学附属医院临床分离.
培养基:肉汤MH(B).对照药:氨苄西林(Ampicillin).
-
MIC测定,细菌培养基的制备:MH(B)营养肉汤分装后于121 ℃高压蒸汽灭菌锅中灭菌20min.实验菌液的制备:以比浊管调整菌液浓度为1×106CFU/mL.微量稀释法:参照NCCLS标准采用微量稀释法进行.生物安全柜使用前紫外灭菌30min.在96孔板第1行中用移液枪加入20 μL药液与180 μL培养基MH(B)并混匀,2~6行每孔加入100 μL,从第1行中吸取100 μL加入第2行并混匀,以此类推,加到第6行,最后吸取100 μL打到废液中,得到2,4,8,16,32,64倍稀释的药物.第7行每孔加入氨苄西林10 μL+90 μL培养基作为阳性对照,第8行直接加入菌液作为阴性对照,同时设立药物稀释浓度梯度和培养基作为空白对照.加入菌液100 μL,37 ℃培养24 h后取出观察.将96孔板放在黑色背衬下观察,孔底呈圆点样的混浊沉淀为细菌生长,孔内液体不显混浊疑似细菌生长被抑制,每个化合物及阳性对照重复3次.实验操作见参考文献[23].
-
细胞及来源:人肺癌细胞A549由贵州医科大学药学院本课题组前期保存;人结肠癌细胞HT29购于国家实验细胞资源共享服务平台;人肾小管正常细胞HK2购于上海复祥生物科技有限公司.
培养基:RPMI-1640培养基(10%胎牛血清),DMEM/F12培养基(10%血清).
对照药:辛二酰苯胺异羟肟酸(SAHA).
-
按参考文献[24]采用MTS比色法对目标化合物A1-A28进行体外抗肿瘤活性测试.贴壁细胞用0.25%胰蛋白酶消化传代.将处于对数生长期的细胞制成单细胞悬液,调整细胞数为5×104~1×105个/mL加于96孔培养板中,每孔100 μL,6个复孔,周围用PBS填充提供水分,于37 ℃,5% CO2培养箱中培养. 12~24 h后,待细胞贴壁后,吸走旧的培养基,将待测化合物用DMSO溶解成所需的浓度,以培养基(10%胎牛血清)稀释成50 μmol/L,25 μmol/L,12.5 μmol/L,6.25 μmol/L,3.13 μmol/L,1.56 μmol/L的终浓度加入到相应的孔板中,每孔100 μL,并设置阳性对照与空白对照组(未经化合物处理的培养基).置于37 ℃,5%CO2培养箱中培养48 h后,每孔加入5 μL MTS,继续于培养箱中培养3 h后,将96孔板于酶标仪(波长为490nm)中测试吸光度值(OD),计算细胞抑制率及半抑制浓度IC50,实验重复3次.细胞抑制率计算公式如下:
空白组:即未经化合物或阳性对照药处理;加药组:经化合物或阳性对照药处理.
2.1. 细菌活性测试
2.1.1. 实验材料
2.1.2. 实验操作
2.2. 抗癌活性测试
2.2.1. 实验材料
2.2.2. 实验操作
-
结合前期工作[7, 9]及按照文献[9]方法,我们采用HBTU缩合剂,DIPEA为相转移催化剂,CH2Cl2为反应溶剂,在室温下搅拌反应24 h,产物A1收率仅为28.5%.可能是杂环羧酸酰化活性低、中间体4亲核力弱及空间位阻大等原因,加之反应时间长,副产物多,后处理及分离纯化十分困难.为了缩短反应时间,提高产物收率,我们以合成A1为例,考察不同催化剂及其用量,反应溶剂、温度、时间等参数对产物收率的影响,优化其合成条件.各反应参数对A1收率的影响如下:
-
在原料比为1:1(mmol),设置反应温度50 ℃,反应时间为2 h,采用催化剂(C4H9)4NBr,溶剂5mL CH3CN,分别在催化剂与中间体4a(mmol)用量比为0.5:1,1.0:1,2.0:1,2.5:1的条件下制备目标产物A1.发现用量比为2.0:1和2.5:1时A1收率相对较高.为了提高产物收率,我们进一步考察了不同催化剂DIPEA,DCC,DMAP对A1收率的影响,实验结果见表 2.
结果表明,DIPEA的催化效果最好,当用量比增至2.5:1时,产物收率增大的幅度极小,而DIPEA浪费较大,且过多DIPEA存在反应体系中不利于目标物的分离纯化,因此选择用量比为2.0:1.
-
在室温下,以催化剂DIPEA与中间体4a用量比为2:1,原料比为1:1(mmol)的条件下,分别溶于二氯甲烷(CH2Cl2)、四氢呋喃(THF)、乙腈(CH3COCH3)、丙酮(CH3CN)、甲苯(C6H5CH3)溶剂中搅拌反应体系2 h,TLC跟踪反应几乎无产物点.再分别保持各反应体系微沸腾回流并搅拌反应体系2 h制备目标产物,经分离纯化处理后,发现以THF为反应溶剂A1收率相对较高(50.8%).由此,我们进一步考察反应时间对A1收率的影响,实验结果见表 3.
从表 3中看出:在THF(66 ℃)反应溶剂中,反应时间由2 h至7 h,A1收率从50.8%增加到80.4%,同时结合TLC显示,反应时间在6 h时副产物明显增加,不利于后处理纯化;延长反应至7 h产率反而降低,因此,确定5 h为最佳反应时间.同时,我们还重点考察对比5 h反应时间内不同溶剂回流状态及干燥THF溶液中A1收率对比;以及5~7 h内CH2Cl2与THF溶剂中A1收率对比.实验结果表明:用干燥处理的THF为反应溶剂,保持反应体系微沸腾(66 ℃)回流搅拌5 h获得A1收率为82.3%.
-
以氨苄西林(Ampicillin)为阳性对照药,采用二倍稀释法对大肠杆菌(E.coli)、铜绿假单胞杆菌(P.aeruginosa)、金黄色葡萄球菌(S.aureus)及多重耐药菌:耐药大肠埃希菌(N-E.coli)、耐药性铜绿假单胞杆菌(N-P.aeruginosa)、耐甲氧西林金黄色葡萄球菌(MRSA)进行最低抑菌浓度MIC测试,实验结果见表 4.
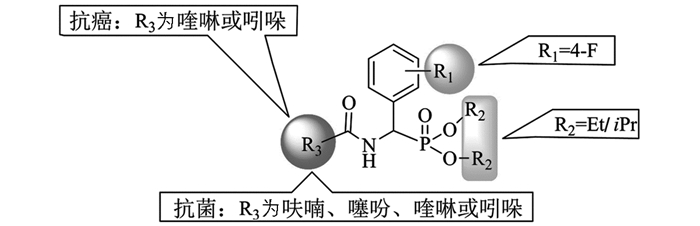
由表 4可知,部分化合物具有较好的抗菌活性,尤其对大肠杆菌(E.coli)及铜绿假单胞杆菌(P.aeruginosa)的抑制活性较明显,其中化合物A4,A8,A22,A24,A27,A28对E.coli的MIC分别为1,1,2,1,2,0.25mg/mL,与对照药氨苄西林(MIC=0.1mg/mL)接近或相当;A4,A22,A27,A28对P.aeruginosa的MIC分别为2,1,1,0.5 mg/mL,接近对照药的MIC.相比之下,大部分化合物对金黄色葡萄球菌(S.aureus)和耐药菌不敏感,但化合物A27对耐甲氧西林金黄色葡萄球菌(MRSA)抑制效果较好,其MIC为4mg/mL,与对照药氨苄西林(MIC为2mg/mL)相当,其次是化合物A28对耐药性大肠杆菌(N-E.coli)、耐药性铜绿假单胞杆菌(N-P.aeruginosa)的MIC分别为8 mg/mL,4 mg/mL(阳性对照药分别为2,1 mg/mL).
-
以SAHA为阳性对照药,采用MTS比色法对人肺癌细胞(A549),人结肠癌细胞(HT29)以及人肾小管正常细胞(HK2)进行体外抗癌活性测试,以化合物及阳性对照药(SAHA)浓度均为50 μmol/L进行初筛,实验结果见表 5.
由表 5可知,部分化合物具有由中等至好的抗癌活性,其中化合物A16(R1=-F,R2=-iPr,R3=6-甲-3吡啶)抗肿瘤HT29的抑制率为73.9%;化合物A28(R1=-F,R2=-iPr,R3=(1H)-3-吲哚)抗肿瘤A549,HT29的抑制率分别为76.9%和69.6%,与对照药SAHA(抑制率81.6%,82.8%)接近;尤其是A24(R1=-F,R2=-iPr,R3=6-喹啉)抗HT29活性明显,抑制率为78.1%,与对照药抑制率接近.而化合物对人肾小管正常细胞(HK2)的抑制率为11.1%~28.5%.而对照药SAHA作用于HK2时存活率仅为22.8%,相比较之下,对照药SAHA对正常细胞(HK2)的损伤程度更大.整体来看,大多数化合物的抗肿瘤活性较差,但少数化合物对特定的肿瘤细胞显示不错的活性.
我们进一步测试了化合物A16,A24和A28的IC50值,结果见表 6.实验结果表明,化合物A28对A549的抑制活性较好,其IC50值为17.1 μmol/L(对照药IC50为12.5 μmol/L);化合物A24对HT29的IC50为18.0 μmol/L,与对照药IC50为14.1 μmol/L接近.化合物A24,A28对HK2IC50值均超过120 μmol/L(对照药IC50为33.7 μmol/L),说明化合物对癌细胞有较好的抑制活性,并对人肾小管正常细胞的毒性较小.
-
从化合物结构与活性数据关系(SAR)分析来看(图 3),当取代基R1=F,R2=Et或iPr,R3为呋喃、噻吩、喹啉或吲哚杂环时,大部分化合物抑制E.coli和P.aeruginosa的活性较明显.此外,当R1,R2不变,R3为喹啉或吲哚杂环时,部分化合物对肿瘤细胞A549和HT29的抑制活性较好.因此,对该类化合物进一步的结构改造与优化,为获得新型杂环膦酸酯抗菌、抗癌化合物单体,具有重要的理论和实验意义.
3.1. 合成条件优化
3.1.1. 催化剂及其用量对A1收率的影响
3.1.2. 溶剂、反应温度及反应时间对A1收率的影响
3.2. 活性测试结果与讨论
3.2.1. 抗菌活性
3.2.2. 抗癌活性
3.2.3. 结构与活性关系(SAR)
-
通过反应条件优化合成了一系列新型杂环磷酸酯衍生物A1-A28,并测试了目标物生物活性.实验结果表明,部分化合物具有较好的抗菌、抗癌活性,如化合物A4,A22,A27,A28抗E.coli和P.aeruginosa抑制率与对照药氨苄西林(MIC=0.1mg/mL)接近或相当,尤其是化合物A27抗耐甲氧西林金黄色葡萄球菌(MRSA)抑制效果明显.同时,化合物A24和A28抗肿瘤活性也明显,A24对HT29的IC50与对照药接近,A28对A549的IC50与对照药相当.值得注意的是,所有化合物对人肾小管正常细胞(HK2)的存活率超过70%,相比对正常细胞(HK2)的毒性弱于对照药SAHA,具有进一步的研究意义.