




-
猪传染性胃肠炎(porcine transmissible gastroenteritis,TGE)是由猪传染性胃肠炎病毒(porcine transmissible gastroenteritis virus,TGEV)引起的一种以严重腹泻、呕吐和脱水为临床症状的高度胃肠道传染性疾病[1-2],尤其对2周龄以内的仔猪有很高的致死率,据报道死亡率可达100%[3].该病最早发现于美国,目前呈世界性分布,给养猪业造成了巨大的损失. TGEV属冠状病毒科冠状病毒属、基因组不分节段的单股正链RNA,长约2.86×104 bp[4-5].其主要包括4种结构蛋白:M蛋白、E蛋白、S蛋白和N蛋白[4].其中M基因是一个重要的编码基因,全长789 bp,可编码262个氨基酸,不同毒株间M蛋白具有高度保守性.该蛋白从N端到C端可分为信号肽、膜外区、跨膜区、歧性区及突出于病毒粒子的C端区5个功能域,主要包埋在病毒脂质囊膜中,在病毒颗粒装配过程中将核衣壳连接到囊膜上.冠状病毒M蛋白在病毒粒子的组装和出芽中起最重要的作用[6-7],但M蛋白自身不能够出芽,必须与sM蛋白相结合[8]. M蛋白是一种3次跨越囊膜的完整膜糖蛋白,是冠状病毒最丰富的囊膜蛋白[9-10]. M蛋白不仅影响病毒的变异,并局限于内质网膜上决定病毒的装配位点[11],从而在病毒装配期间将核衣壳连接到囊膜上[12];M蛋白能自我相互作用,形成均一的复合体,也能与S,E,N蛋白等作用形成不均一复合体,M蛋白与这些结构蛋白相互作用,使得它们能共同组装成病毒粒子,但是M蛋白必须首先与E蛋白作用才能被装入病毒粒子,单独M蛋白不能诱导病毒样颗粒(VLPs)的形成.
有囊膜的病毒通过出芽的方式从宿主细胞获得囊膜,在此过程中病毒的囊膜蛋白形成1个特殊的膜相结构与其他病毒组分共同协作促使病毒出芽[13].例如,一些病毒包括人HIV以外吐的方式从宿主细胞的质膜出芽和释放病毒颗粒.一些有囊膜的病毒在细胞内膜出芽[14-15].病毒颗粒用细胞内膜结合的隔室包装自己,如内质网、高尔基体及新发现的病毒从宿主细胞出芽的细胞分泌途径[16].然而病毒粒子组装与出芽方式的详细机制,尤其是宿主因子在病毒粒子组装与出芽过程中的作用,在很多病毒的研究中还未有报道.本研究以猪传染性胃肠炎病毒M蛋白构建酵母双杂交的诱饵载体pGBKT7-M,检测其对酵母细胞是否有毒性、自激活活性及在酵母细胞内的表达情况,为了解宿主因子在病毒粒子组装、出芽和成熟过程中的相关作用奠定基础.
HTML
-
含有TGEV M基因全长的pFastBacTM-M质粒由本试验室构建并保存;质粒提取试剂盒购自Genview公司;pMD19-T simple克隆载体、限制性内切酶、T4 DNA ligase及所用Marker购自宝生物工程(大连)有限公司;大肠杆菌DH5α感受态细胞购自北京全式金生物技术有限公司;MatchmakerTM Gold Yeast Two-Hybrid System、酵母转化试剂盒、X-α-Gal、酵母菌株、Aureobasidin A等均购自Clontech公司;各种营养缺陷型培养基、酵母蛋白提取试剂盒购自上海生工生物公司;YNB购自北京索莱宝生物公司;胶回收试剂盒购自OMEGA公司;c-Myc标签抗体购自ProteinTech公司;ECL显色试剂盒购自碧云天生物公司;引物合成和测序由上海生工生物公司完成.
-
根据本试验室构建并保存的含有TGEV M基因全长的pFastBacTMDual-M质粒的序列,设计M基因引物为上游引物:5'-CCGGAATTCATGCGCTATTGTGCTATGAAAT-3',其5'端下划线部分为限制性酶切位点EcoRⅠ;下游引物:5'-GGGGGATCCTTATACCATATGTAATAATTTTTCTTG-3',其5'端下划线部分为限制性酶切位点BamHⅠ.预期扩增片段大小为738 bp,为M基因去除信号肽片段大小.
PCR反应体系如下:2×Taq PCR Master Mix 25 μL,上、下游引物(20 μmol/L)各0.5 μL,pFastBacTMDual-M质粒2 μL,ddH2O 22 μL.
PCR反应条件:94 ℃ 3 min,94 ℃ 30 s,58 ℃ 30 s,72 ℃ 1 min,33个循环,72 ℃ 10 min. PCR扩增产物经1.0%琼脂糖凝胶电泳鉴定,胶回收试剂盒对目的条带进行回收纯化,取3 μL回收产物电泳检测胶回收纯化是否成功.
-
将胶回收纯化后的PCR产物与克隆载体pMD19-T Simple进行连接,反应体系为pMD19-T Simple vector 1 μL,M基因胶回收产物3 μL,ddH2O 1 μL,solutionⅠ5 μL,16 ℃连接4 h.将连接产物转化成DH5α感受态细胞,涂布于含Amp的LB平板上进行蓝白斑筛选,37 ℃倒置培养8~12 h.随机挑取单个阳性克隆摇床培养8~12 h,参照质粒提取试剂盒提取重组质粒,经PCR反应、EcoRⅠ/BamHⅠ双酶切验证,且对阳性质粒进行测序鉴定,鉴定正确的重组质粒命名为pMD19-T Simple-M.
-
将阳性质粒pMD19-T Simple-M及诱饵载体pGBKT7分别进行EcoRⅠ/BamHⅠ双酶切反应,胶回收纯化后使用T4 DNA连接酶将双酶切后的M基因片段与pGBKT7 16 ℃连接10 h.连接产物转化大肠杆菌DH5α感受态细胞,涂布于含Kan抗性的LB固体平板上,37 ℃倒置培养8~12 h,随机挑取单菌落到含Kan抗性的LB液体培养基中,37 ℃摇床培养8~12 h,提取重组质粒经PCR反应及双酶切验证,并经过阳性质粒测序鉴定,鉴定正确的重组质粒命名为pGBKT7-M.
-
采用TE/LiAc法制备酵母菌感受态细胞,将酵母菌Y2HGold从-80 ℃冰箱取出,划线接种至固体YPDA平板上,30 ℃倒置培养3~5 d,挑取1个2~3 mm的单菌落接种于含3 mL液体YPDA培养基上,230 r/min 30 ℃摇床培养8~12 h,取5 μL培养物于50 mLYPDA培养基中,230 r/min 30 ℃摇床培养至OD600 nm=0.15~0.3. 2 450 r/min离心5 min,弃上清,用新鲜的YPDA培养基100 mL重悬细胞沉淀,230 r/min 30 ℃摇床培养至OD600 nm=0.4~0.5,将培养物分至两个50 mL管中,2 450 r/min离心5 min,弃上清,每管用30 mL灭菌去离子水重悬细胞沉淀,2 450 r/min离心5 min,弃上清,每管沉淀用1.5 mL 1.1×TE/LiAc重悬,转移细胞悬液于2个1.5 mL灭菌管中,高速离心15 s,再分别用600 μL 1.1×TE/LiAc重悬细胞沉淀,此时感受态细胞已制备完成.
在预冷的1.5 mL灭菌管中分别加入pGBKT7-M重组质粒和pGBKT7空质粒100 ng,经变性处理后的Yeastmaker Carrier DNA 5 μL,感受态细胞60 μL,轻柔地混匀后加入新鲜配制的PEG/LiAc 500 μL,轻柔地混匀,30 ℃温箱内静置孵育50 min(每10 min轻柔地颠倒混匀1次),加入20 μL DMSO混匀后42 ℃水浴15 min(每5 min轻柔地混匀1次),2 450 r/min离心5 min后弃上清,用1 mL YPD plus Medium重悬细胞沉淀,220 r/min 30 ℃摇床培养50 min,高速离心15 s后弃上清,使用1 mL 0.9%(W/V)NaCl溶液重悬细胞沉淀.将转化产物1:10稀释后取100 μL涂布于SD/-Trp营养缺陷培养基,30 ℃培养箱内倒置培养3~5 d.
-
将空载体pGBKT7和诱饵载体pGBKT7-M分别转化至酵母菌Y2HGold感受态细胞,转化产物分别按1:10,1:100进行稀释,各取100 μL涂布于SD/-Trp平板,30 ℃倒置培养3~5 d.从平板上随机挑取直径为2~3 mm单菌落接种到SD/-Trp液体培养基中培养,220 r/min 30 ℃摇床培养20~24 h后测OD600 nm值.以空载体pGBKT7为对照,对比诱饵载体pGBKT7-M平板上菌落的数量及菌落大小,评价构建的诱饵载体对酵母菌株的毒性作用.
-
将诱饵载体pGBKT7-M转化至Y2HGold酵母感受态细胞,转化产物按1:10,1:100进行稀释后,分别涂布100 μL到SD/-Trp,SD/-Trp/X-α-Gal,SD/-Trp/X-α-Gal/AbA平板上,30 ℃恒温箱内倒置培养3~5 d,观察比较各平板上菌落生长情况.
-
参照酵母蛋白提取试剂盒提取诱饵蛋白,SDS-PAGE电泳后转膜,5%脱脂奶粉常温封闭4 h,加入一抗c-Myc标签抗体4 ℃孵育8~12 h,洗涤3次,每次10 min后再加入二抗常温孵育2 h,ECL显色.
1.1. 材料
1.2. 方法
1.2.1. M基因的扩增、回收及鉴定
1.2.2. pMD19-T Simple-M重组质粒的构建及鉴定
1.2.3. 诱饵质粒pGBKT7-M的构建及鉴定
1.2.4. 酵母感受态细胞的制备及转化
1.2.5. 诱饵质粒pGBKT7-M毒性检测
1.2.6. 诱饵载体pGBKT7-M自激活活性的检测
1.2.7. 诱饵蛋白在酵母中的表达验证
-
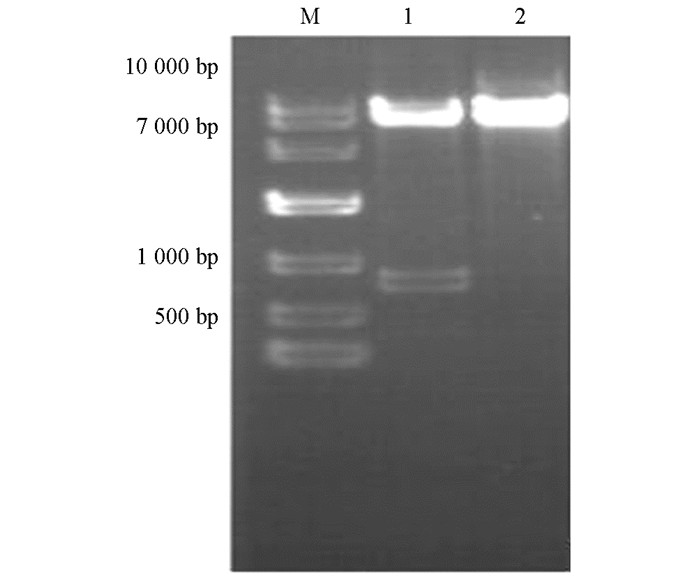
提取重组质粒,经EcoRⅠ/BamHⅠ双酶切验证,pGBKT7大小为7 300 bp,M基因为738 bp,电泳结果显示符合目的条带大小基本一致(图 1),且测序结果显示碱基序列未发生突变与缺失.
-
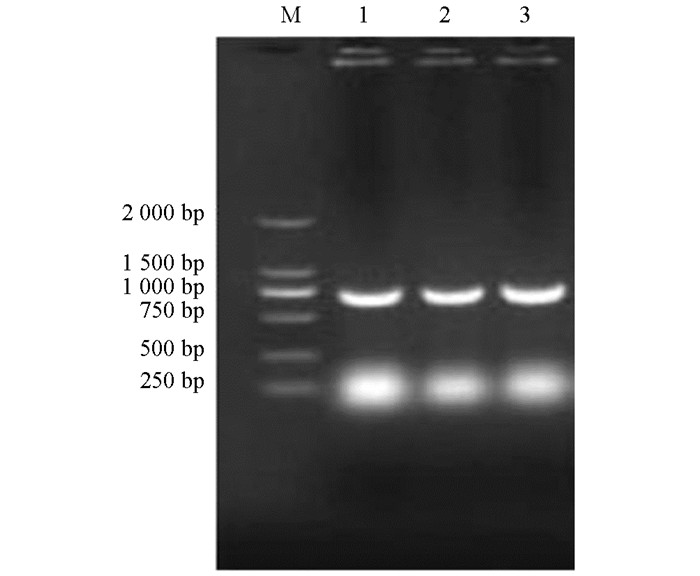
挑取选择培养基SD/-Trp平板上长出的单菌落进行菌落PCR,经1%琼脂糖电泳后,所得目的条带738 bp左右,与预期大小相符(图 2).
-
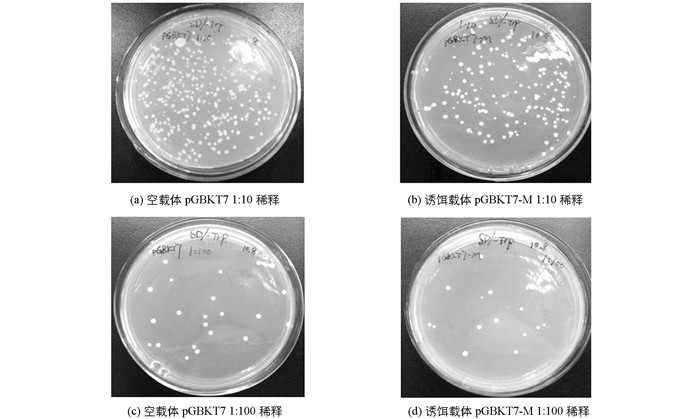
将空载体pGBKT7和构建的诱饵载体pGBKT7-M分别转化至Y2HGold酵母感受态细胞中,对选择性培养基SD/-Trp平板上长出的菌落数量及菌落大小进行差异对比,结果证明空载体和诱饵载体的菌落大小基本一致,诱饵载体pGBKT7-M上的菌落数量略少于空载体pGBKT7(图 3),统计学分析无明显差异,说明诱饵载体pGBKT7-M对酵母Y2HGold细胞没有明显毒性.
-
将诱饵载体pGBKT7-M转化至酵母Y2HGold感受态细胞中,转化产物按1:10,1:100进行稀释后,分别涂布于选择性培养基SD/-Trp,SD/-Trp/X-α-Gal,SD/-Trp/X-α-Gal/AbA平板上进行培养,可观察到在SD/-Trp平板上为白色菌落,SD/-Trp/X-α-Gal平板上为蓝色菌落,SD/-Trp/X-α-Gal/AbA平板上没有菌落生长(图 4).
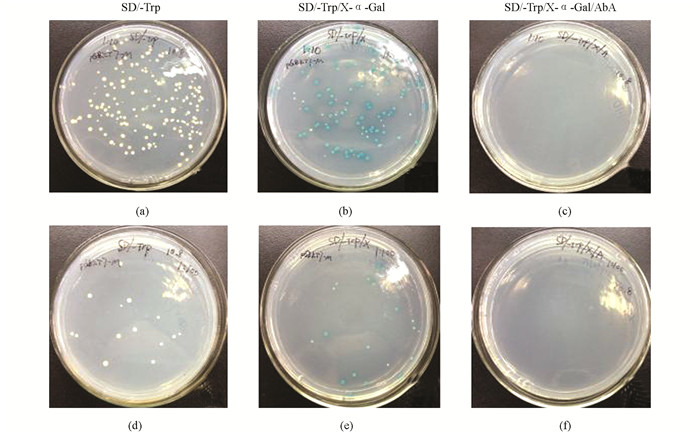
-
提取pGBKT7-M酵母蛋白,经Western blot分析可见融合蛋白表达,蛋白大小约为5×104,阳性对照pGBKT7-53蛋白大小约为5.7×104,与预期结果一致,说明诱饵载体在酵母细胞Y2HGold中正确表达(图 5).
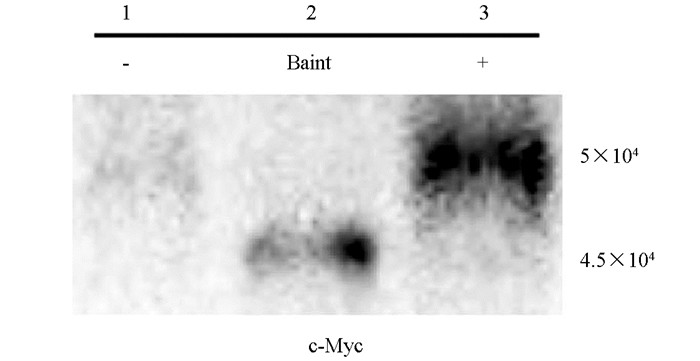
2.1. 诱饵载体pGBKT7-M的酶切鉴定
2.2. 诱饵载体转化酵母菌Y2HGold的鉴定
2.3. 诱饵表达载体pGBKT7-M的毒性检测
2.4. 诱饵载体pGBKT7-M的自激活检测
2.5. 诱饵蛋白的表达检测
-
蛋白-蛋白相互作用对执行各种生物功能、病毒与宿主互作方面具有极其重要的作用,蛋白间互作的机制将有助于对已知全长基因但未知功能基因的功能分析[17].在蛋白质相互作用的研究方法中,酵母双杂交系统(Y2H)是最常见的蛋白互作方法[18],由于此方法巧妙地把研究复杂蛋白质之间的相互作用转换成易操作的核酸研究,并能进行高通量的蛋白筛选,为研究蛋白质之间的相互作用提供了一条捷径.自从1989年由Fields等[19]在进行真核基因转录调控研究中用来检测酵母体内蛋白与蛋白相互作用的系统,这种研究蛋白质相互作用的试验方法已得到越来越广泛的应用.
为筛选TGEV M蛋白与宿主细胞猪小肠上皮细胞中相互作用的蛋白,本试验成功构建了用于酵母双杂交的诱饵载体pGBKT7-M.表达载体pGBKT7含有色氨酸编码基因及Kan抗性筛选基因,故构建的诱饵质粒pGBKT7-M能在SD/-Trp/Kan(50 μg/mL)平板上正常生长,菌落的大小和生长速度与空载体pGBKT7无显著差异,表明诱饵载体pGBKT7-M无毒性.诱饵载体pGBKT7-M在SD/-Trp/Kan平板上正常生长白色菌落,SD/-Trp/X-α-Gal/AbA平板上无菌落长出,表明构建的诱饵载体pGBKT7-M无自激活活性.因此,构建的重组诱饵载体pGBKT7-M可用于酵母双杂交系统筛选与M蛋白相互作用的宿主蛋白.
病毒感染宿主以吸附到宿主细胞表面开始,之后将核酸注入到宿主细胞,并在其中大量繁殖,该过程需要宿主细胞膜上的病毒受体及细胞中的相关蛋白辅助完成[20].因此,研究病毒与宿主细胞的相互作用对阐明TGEV的感染机制有重要意义.关于TGEV病毒的致病机理、与宿主相互作用机制的可用信息相对较少,当前的研究主要集中在病毒基因组结构和功能的研究[21],宿主细胞病毒受体的发现将有助于揭示病毒与机体相互作用的本质,从而了解病毒的致病机制和免疫机理[22].本试验构建的诱饵质粒pGBKT7-M符合筛选酵母双杂交宿主细胞cDNA文库的需要,为进一步筛选与M蛋白相互作用的蛋白奠定了一定的基础,有助于揭示病毒的分子致病机制、与宿主细胞的相互作用机制和寻找病毒的作用靶标,为病毒的预防诊治提供理论依据和解决途径[23].
 DownLoad:
DownLoad: