




-
开放科学(资源服务)标志码(OSID):

-
甘蓝型油菜分枝角度主要指有效分枝与主茎之间形成的夹角[1]. 分枝角度是检测植物空间分布能力的一个重要指标,受遗传因素、植物激素和环境因素等多重因素调控[2-4]. 甘蓝型油菜株型高大、分枝多且长,而分枝角度太大会造成分枝间相互交叉缠绕,降低机械收获时的分行、切割和输送效率[5],油菜机械化收获产量的取得得益于小的分枝角度[6]. 王俊生等[7]提出理想油菜株型应该包括成株期分枝角度较小等一系列特征. 曾川等[8]发现油菜株型在开花期和角果形成期应表现为分枝紧凑. 陈新军等[9]研究发现分枝角度小的紧凑型品种更抗倒伏,单株生产力较高,同时机械化收获产量也较高. 李扬等[10]提出分枝角度的大小决定油菜冠层结构的紧凑度,而紧凑度适宜的株型既可以提高种植密度,也可以减少机械化收获时因植株分枝间的缠绕造成的产量损失. 张倩[11]发现株型紧凑的油菜更适宜密植,对群体的产量和抗倒伏性贡献率较高.
目前,对于油菜分枝角度基因定位的研究还较少. 张倩[11]对分枝形态差异较大的两个亲本F2后代进行研究,检测到1个位于LG1连锁群上的分枝角度数量性状座位(Quantitative Trait Locus,QTL),该QTL分布在标记SWUC893和SWUC816b之间. 段秀建[12]在研究油菜自然群体的分枝角度时发现1个候选基因,该基因与拟南芥基因AT5G14090(AtLAZY1)同源,且该基因的调节机制主要是重新分布植物内的生长素,从而调节腋芽的向地性,达到改变分枝角度大小的作用. 沈钰森[13]利用小孢子培养得到的DH群体进行遗传分析,在全基因组范围内检测到17个与分枝角度性状相关的QTL,其中位于A03上的1个QTL和C03上的2个QTL为新发现的主效QTL,检测到SAUR30(BnaC03g14890D)和SAUR55(BnaC03g16420D)可能与生长素早期响应和油菜分枝角度大小的调控相关. 孙程明[14]利用Illumina 60K SNP芯片对520份甘蓝型油菜材料进行基因分型,联合使用3种模型,共检测到56个显著位点,预测到77个候选基因. 汪文祥等[15]以小孢子培养得到的DH群体为实验材料,利用油菜60K SNP芯片对DH群体基因分型,定位到17个分枝角度QTL,并获得1个与分枝角度相关的候选基因VAMP714,Wang等[16]采用集团分离分析法(BSA)结合下一代测序技术对分枝角度QTL进行精细绘制,鉴定出一个位于A06染色体上的主效QTL,并在该QTL区域鉴定出多个基因,其中BnaA0639380D可能是油菜分枝角度的候选基因.
本文以分枝角度大的纯系黄花亲本62与分枝角度小的纯系粉花亲本77为亲本构建114个DH群体株系,通过GBS简化测序[17]获得SNP数据,考察DH群体一次分枝角度表型,利用一般线性模型(General Linear Model,GLM)[18]进行全基因组关联分析.
HTML
-
黄花亲本62和粉花亲本77都是小孢子加倍后的纯系材料. DH群体为(黄花62×粉花77)F1花粉小孢子加倍成功的114个基因型株系.
-
2018年9月-2019年5月,将亲本和DH群体种植于重庆市北碚区歇马镇西南大学油菜生物学团队育种基地. 每个材料种植3行,每行8株,行距40 cm,株距30 cm,田间管理按常规方式进行. 2个亲本材料分别为62-1,77-1. 62-1为分枝角度大的亲本,平均一次有效分枝角度为43.4°. 77-1为分枝角度小的亲本,平均一次有效分枝角度为38.0°. 油菜主茎上的一次分枝角度和主茎的夹角直接用量角器测量. 分枝角度(BT)在群体中每个单株测5次夹角值,选长势一致的5个单株进行测量.
-
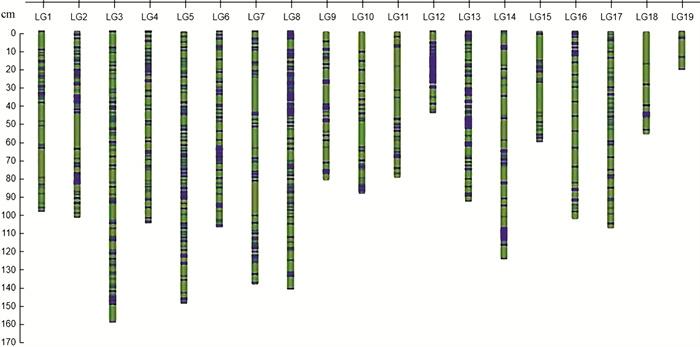
2018年12月,采集亲本和DH群体植株嫩叶,20℃保存备用. 2019年1月送北京诺禾致源生物信息科技有限公司测序. 2个亲本DNA建库类型为DNA-350 bp,以Illumina HiSeq PE150方法测序,测序得到的总碱基数与待测基因组大小的比值即测序深度,该数值为30×[19-20]. DH子代进行GBS简化基因组测序. 利用GBS-SNP-CROP[13]分析流程通过barcode文件得到114个DH子代系的双末端测序文件. 以法国甘蓝型油菜Darmor-bzh为参考组,采用BWA-mem软件比对测序reads到参考组,并通过SAMtools[21]软件参数mpileup获得114个DH子代系的SNP数据.
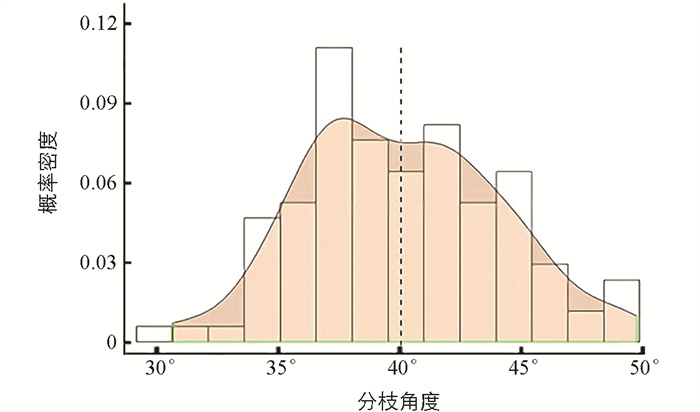
用Excel 2019计算出每个性状的平均值,通过R3.5.0软件包画出分枝角度频数分布直方图和概率密度曲线图(图 1). 用SPSS统计分析软件计算出数据的统计信息,并用Kolmogorov-Smirnov[21]方法验证数据的正态分布.
-
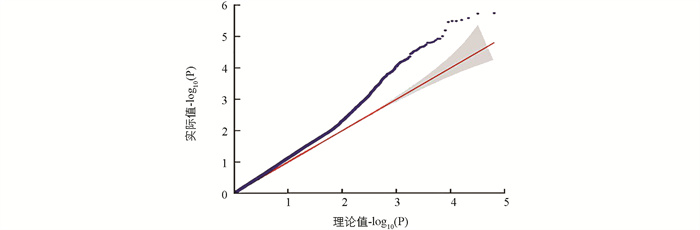
利用TASSEL关联分析软件[22]中的一般线性模型,结合基因型数据,进行全基因组关联分析,以每个SNP位点的-log10(P)观察值和期望值,绘制Quantile-Quantile散点图(QQ Plot)和曼哈顿图(Manhattan Plot). 在Manhattan Plot中,SNP位点P的阈值小于2.59×10-6,表示位于该阈值以下的SNP位点与性状关联程度较强.
-
KEGG采用R3.6.0/clusterProfiler包,在所定位的油菜QTL区间中,找到与油菜基因同源的拟南芥基因,利用R包中自带的拟南芥数据库,通过KEGG富集出可能参与的代谢通路.
1.1. 材料
1.2. 方法
1.3. DH群体GBS测序和数据处理
1.4. 全基因组关联分析(GWAS)
1.5. KEGG富集
-
DH群体一次分枝角度频数分布直方图和概率密度曲线如图 1所示. 利用统计学Kolmogorov-Smirnov方法检测到分枝角度性状(BT)服从正态分布(表 1),表明一次分枝角度可能受主效基因控制,因此对分枝角度性状进行主效基因定位分析.
-
利用DH群体GBS简化测序获得的SNP数据(图 2)和一次分枝角度表型数据进行GWAS定位分析. QQ图(图 3)表明,GLM模型检测到的P值基本接近期望值,具有较好的控制假阳性能力. 利用GLM模型对分枝角度进行关联定位,Manhattan图展示分析结果(图 4),当设置显著性阈值-log10(P)=6.5,共检测到8个显著位点. 这8个显著位点都分布在A07染色体(表 2). 其中显著性最高位点为3469428A07和3469437A07,-log10(P)为8.08,对表型变异的贡献率都为30.72%. 其余位点的-log10(P)值范围是7.17~7.56,可解释25.21%~35.05%的表型变异.

-
依据法国甘蓝型油菜参考组序列信息,8个显著的SNP位点区间内共有49个基因(表 3). 结合法国甘蓝型油菜基因序列和拟南芥同源基因序列注释这49个基因. BnaA07g01910D和BnaA07g01920D基因与拟南芥基因AT5G13370和AT5G13360序列或结构域高度同源,而拟南芥基因AT5G13370和AT5G13360编码与生长素合成相关的GH3家族蛋白. 已有多个文献研究结论推测分枝角度和生长素密切相关,本研究在GWAS定位分枝角度A07染色体区间内,令人意外地注释到了与生长素合成的油菜基因.
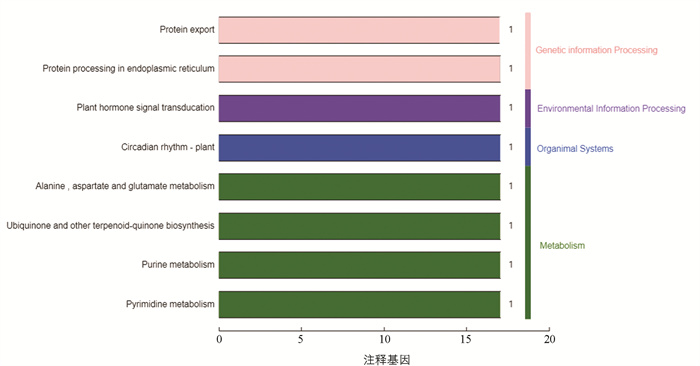
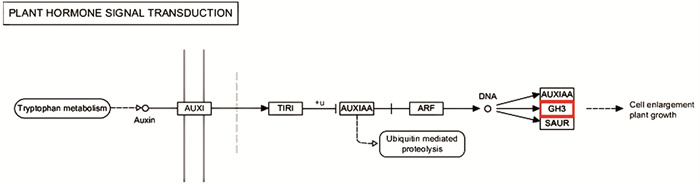
继续对定位区间内的49个基因进行KEGG分析,本研究发现8个基因分别富集到8个代谢通路上(表 4). BnaA07g03650D在嘌呤代谢通路上,BnaA07g01280D参与泛醌及其他萜类醌的生物合成,BnaA07g05610D参与蛋白转运,BnaA07g01250D参与生理周期调节,BnaA07g05090D参与嘧啶代谢,BnaA07g01910D在植物激素信号转导中起作用,BnaA07g05610D参与内质网中的蛋白质加工,BnaA07g03650D参与丙氨酸、天冬氨酸和谷氨酸代谢,而BnaA07g01920D却没有富集到任何通路上,其余40个甘蓝型油菜基因也未被富集到任何代谢通路上(图 5). 本研究在KEGG网站上查询ko04075通路,发现BnaA07g01910D参与的GH3家族蛋白负责细胞繁殖扩增的功能(图 6).
2.1. 一次分枝角度表型分析
2.2. 一次分枝角度GWAS分析
2.3. 一次分枝角度候选基因挖掘
-
甘蓝型油菜第一次分枝角度与遗传基因密切相关. 李洪戈[23]、张倩[11]、汪文祥等[15]利用F2分离群体检测到甘蓝型油菜一次分枝角度由一对主效基因和多个微效基因共同控制. 本研究考察DH群体一次分枝角度表型以及利用简化测序SNP数据对此表型进行GWAS分析,显示出一次分枝角度表型为一对主效基因的特点,且一次分枝角度表型基因效应与以往学者的研究结论基本一致.
本研究通过GWAS分析流程检测到8个显著的SNP位点与一次分枝角度密切关联. 这8个SNP位点分别位于A07染色体1.5Mb,0.9Mb,3.4Mb,6.1Mb附近,此区间内分布有49个基因. 从传统QTL分析思路上可认为,一次分枝角度在A07染色体上端部位有3个主效QTL. 而张倩[11]检测到A01染色体有1个分枝角度QTL,沈钰森[13]发现位于A03上的1个QTL和C03上的2个QTL与分枝角度高度关联. 孙程明[14]定位到52个显著SNP位点与分枝角度相关,汪文祥等[15]定位到17个分枝角度QTL,分析推测C08染色体上1个QTL可能控制甘蓝型油菜分枝角度. 因此,不同作者使用不同群体利用不同分析方法得到的甘蓝型油菜分枝角度主效基因定位和QTL定位结论差异很大. 原因在于分枝角度是一个很复杂的生物学现象,不仅受遗传基因作用,还受激素和环境因素的影响.
GWAS和传统QTL本质上都是寻找标记和性状之间的关联,但GWAS与传统QTL相比较具有3个优势[24]. ①分辨率高. 基于家系群体如BC1、F2、RIL都是先计算Marker之间的重组率,再计算遗传距离,从而推测Marker与性状之间的关联. 家系群体内发生的重组交换有限,重组事件少,在遗传过程中目标区块没有被完全打碎,重组率分辨度不够导致鉴定的关联候选区域特别大. 而GWAS分析的自然群体内部随机交配多,能够把连锁区块打得足够碎,重组事件足够多,定位到的区间足够小,因此分辨率更高、更精确. ②不需要构建家系群体,节约时间和成本. BC1和F2群体至少需要2年,RIL群体需要5年以上,而自然群体或DH群体需要年限短,且群体内部保留了大量的遗传变异位点. ③能识别更多的等位基因. 传统QTL连锁分析针对双亲之间的差异目标性状,家系群体只能分析一个性状,而GWAS一次可以采集群体的多个性状同时进行分析.
本研究所用的SNP位点信息来源于GBS简化测序. 简化测序和全基因组重测序比较有很多不足. 简化测序是酶切全基因组,对酶切位点附件的片段进行变异检测. 酶的选择、酶切效率、酶切位点在物种内的实际分布都影响着SNP数据的收集. 简化测序一般只能鉴定到几万个至10几万个SNP,而全基因组重测序能鉴定到10 Mb以上的SNP数据量[25]. 因此,在经费充足的情况下,最理想的GWAS分析是对群体株系进行全基因组重测序,才能确保染色体上分布足够密度的SNP位点.
本研究DH群体的一次分枝角度表型呈正态分布. GWAS方法将一次分枝角度主效基因定位在A07染色体上. 定位区间内分析和注释了49个基因,KEGG分析推测甘蓝型油菜一次分枝角度主效基因可能为编码GH3家族蛋白的BnaA07g01910D基因,此基因负责细胞繁殖扩增,影响分枝角度表型.
 DownLoad:
DownLoad: