




-
开放科学(资源服务)标识码(OSID):

-
家蚕(Bombyx mori)是一种重要的经济昆虫,蛋白质是生命的物质基础,对家蚕蛋白质组的研究不仅能发现许多以前未报道过的蛋白,还能解释家蚕的各项生命活动. 对新鉴定蛋白的功能进行研究,对改造家蚕品种、遗传育种等方面都具有重要意义. 随着质谱仪分辨率的不断提高和对家蚕研究的不断深入,人们更想知道家蚕整个生命周期各个组织中的蛋白表达情况,而目前家蚕还没有一份高覆盖率的精细蛋白质谱图. 要想获得高覆盖率的精细谱图,除了要采用先进的超高分辨率质谱仪外,还应该对样品质谱前处理的方法步骤进行改良. 在2014年,Nature连载了2篇人类蛋白质组研究成果[1-2],使研究者对人类编码蛋白有了清晰的认识,对物种蛋白质组的全谱鉴定能为相关物种的研究打下良好的基础. 随着家蚕基因组研究的不断深入[3-8],家蚕的研究也进入了后基因组时代,而蛋白质组是后基因组时代的一个重要内容. 钟伯雄[9]采用双向电泳及蛋白质氨基酸序列分析技术,研究了家蚕不同胚胎发育时期的基因表达情况. 徐豫松等[10]应用双向电泳、同位素标记等方法研究了家蚕5龄期和变态期的脂肪体,从5龄蚕检测到270种蛋白质到5龄96 h检测到400多种蛋白质,为了解脂肪体蛋白质的表达规律奠定了基础. 2009年,Li等[11]首次采用鸟枪法LC-MS/MS整合生物信息学方法研究了家蚕5龄第5 d内分泌系统的脑、神经节和胸腺3个器官,分别鉴定出3 430,2 683和3 395种蛋白. 之后,有学者研究了家蚕的头[12]、胚胎[13]、脂肪体[14]等组织器官的蛋白质组,甚至对之前双向电泳没研究过的组织,如触角[15]、附腺[16]、蜕皮液[17]等都做了蛋白质组研究. 利用不同组织样品不同处理方式的蛋白质组数据,有学者对家蚕蛋白质库进行了优化与整合[18],也使得家蚕的比较蛋白质组学慢慢成熟,基于高分辨率质谱仪家蚕蛋白质组全谱鉴定呼之欲出. 蛋白质组常用的样品处理方法是过滤器辅助旋转方法(FASP)[19],该方法可以有效去除制备样品过程中造成的污染. 基于此法的改进方案也有很多[20],有学者通过分级样品来增加蛋白质的鉴定数量[21-24],也有学者通过改良数据算法来增加蛋白质的鉴定数量[25-27],还有学者通过去除高丰度蛋白来提升鉴定效果[28]. 这些改良方法主要是用人类细胞系或者容易获取且稳定的材料做实验,目前还没有在蚕的组织器官或蚕的细胞系开发蛋白质组技术的报道. 如果想采用超高分辨率质谱仪绘制家蚕高覆盖率的蛋白质精细谱图,就需要优化各项参数,系统评估家蚕蛋白质组样品的特殊性. 本研究基于超高分辨率质谱仪,使用家蚕脂肪体改良质谱前处理方法、优化质谱上机参数、优化数据库和分级策略等方式来提高鉴定蛋白质的数量,旨在为大规模蛋白质组测序奠定基础.
HTML
-
实验中所使用的家蚕脂肪体取自‘大造’,该品种由西南大学前沿交叉学科研究院生物学研究中心基因库提供. 幼虫在室温条件下喂食新鲜桑叶,待蚕生长至5龄第3 d,用镊子夹出脂肪体置于装双蒸水的一次性培养皿中,用滤纸吸干水分后放于1.5 mL离心管中,每支离心管大约装400 μL,-80 ℃冰箱中保存备用.
-
取出两管脂肪体样品,一管倒入预冷的研钵中,加入液氮进行研磨;研磨后转移到新的离心管中,加入约400 μL 8 mol/L尿素(Sigma,51456)溶液. 另一管加入400 μL 8 mol/L尿素后用组织研磨器(生工,F520001-0001)在冰上研磨. 水平震荡器(Tomy,MT-360)第9档振荡5 min;11 500 r/min,25 ℃离心15 min,取上清置于新的离心管中;之后用BCA蛋白质定量试剂盒(Sigma,QPBCA)对样品进行蛋白浓度定量.
-
质谱前处理步骤参考FASP酶解步骤[19],将200 μg样品加入10 kDa超滤管(Millipore,UFC5010BK)中,使用8 mol/L尿素溶液补足200 μL,混匀后11 500 r/min室温离心20 min;加入200 μL 8 mol/L尿素,11 500 r/min室温离心20 min;再用8 mol/L尿素洗2次(超滤管管底溶液快满时,将溶液倒出);加入1 mol/L二硫苏糖醇(Sigma,D9163)溶液(DTT)50 μL,150 μL 8 mol/L尿素溶液,管口用封口膜(Bemis,PM-996)密封置于37 ℃孵育2 h;撕开封口膜,加入15 μL 1 mol/L碘乙酰胺(Sigma,I1149)溶液,混匀后,室温避光孵育1 h,11 500 r/min室温离心20 min;加入200 μL 8 mol/L尿素溶液,11 500 r/min室温离心20 min洗2次;加入200 μL 50 mmol/L碳酸氢铵(Sigma,40867)溶液,11 500 r/min室温离心20 min洗3次;将超滤管底座换新,在超滤管中加入400 μL 10 μg/mL胰蛋白酶(Sigma,T6567)溶液,管口用封口膜密封,37 ℃孵育36 h;撕掉封口膜,11 500 r/min室温离心20 min;在超滤管中加入40 μL 50 mmol/L碳酸氢铵溶液,室温11 500 r/min离心20 min,4 ℃冷冻浓缩至样品完全干燥;用脱盐柱(Thermo Fisher Scientific,87784)脱盐,然后在4 ℃浓缩干燥;向脱盐后冻干的肽段加入100 μL 0.1%甲酸水(Thermo Fisher Scientific,85170)溶液,振荡30 s,室温11 500 r/min离心10 min,吸上清10 μL于上样瓶中.
-
色谱部分:采用Thermo Fisher Scientific的EASY-nLC 1000纳升级流速系统进行样品上样,采用预柱(Thermo Fisher Scientific,164564)串联分析柱(Thermo Fisher Scientific,164555)分离肽段. A相为0.1%甲酸水溶液,B相为0.1%甲酸乙腈(Thermo Fisher Scientific,85174)溶液,上样环吸取2 μL样品溶液,吸样流速为8 μL/min,使用16 μL A相溶液用3.5 μL/min的流速推入分析柱中,检测时用梯度乙腈溶液进行检测,流速为0.25 μL/min,上样时间为60 min. 平衡预柱的体积为10 μL,流速为3.5 μL/min;平衡分析柱的体积为5 μL,流速为0.25 μL/min,用100 μL B相清洗自动上样环.
质谱部分:采用Thermo Fisher Scientific公司Q Exactive质谱仪,检测时间为60 min,喷雾电压为2.3 kV,离子传输管温度为275 ℃. 离子模式为正离子模式,默认电荷为2个,全扫的分辨率为70 000,AGC target为1e6,Maximum IT为20 ms,扫描范围为300~1 800 m/z;二级扫描分辨率为17 500,AGC target为1e5,Maximum IT为60 ms,采用Top 20原则采集数据,动态排除时间设置为30 s. 质谱环境温度为22 ℃,空气湿度为50%.
-
原始数据采用MaxQuant 1.3.0.5搜库,数据库为2016年7月从NCBI(
http://www.ncbi.nlm.nih.gov ) 下载的数据和从SilkDB(http://silkworm.swu.edu.cn/silkdb )下载的共32 835条家蚕蛋白质序列的数据库. 可变修饰选择Oxidation(M)和Acetyl(Protein N-term),酶切位点选择Trypsin/P,最大漏切位点2个,最大带电荷7,Protein FDR为0.01,最小肽段长度为6. 得到的数据去除污染序列数据和反向序列数据后用于分析. -
将1.2和1.3中组织研磨方法制备的肽段全部加入上样瓶中,分别用不同的上样时间和不同的上样体积进行质谱检测. ①上样时间分别为60,90,120,150,180,240 min,上样体积为2 μL;②上样体积分别为1,2,4,6,8,10,12,14,16,18 μL,上样时间为90 min. LC-MS/MS、数据匹配等其他参数如1.4和1.5所述.
-
样品制备:取4管5龄第3 d脂肪体样品,分别加入8 mol/L尿素溶液、4% SDS溶液、8 mol/L尿素100 mmol/L DTT溶液、4% SDS 100 mmol/L DTT溶液,之后采用2D蛋白定量试剂盒(GE Healthcare,80-6483-56)进行蛋白质定量,再经FASP酶解后质谱检测,详细步骤如1.3-1.6所述.
-
样品制备:取5龄第3 d脂肪体样品,加入8 mol/L尿素溶液溶解样品,FASP酶解后采用90 min上样时间和10 μL上样量进行质谱检测,获得6次技术重复数据,详细步骤如1.3-1.5所述.
从网址KAIKObase:
http://sgp.dna.affrc.go.jp/KAIKObase ,Uniprot:http://silkworm.genomics.org.cn ,NCBI:http://www.ncbi.nlm.nih.gov ,SilkDB:http://silkworm.genomics.org.cn ,Silkbase:http://silkbase.ab.a.u-tokyo.ac.jp 中下载最新的家蚕蛋白质序列,将二代数据库进行整合得到Merge库. 序列相似性阈值设置为90%,得到非冗余数据库Streamine库,该库含有21 878种家蚕蛋白质. 采用不同的数据库分别搜索,其他质谱数据匹配参数如1.3-1.5所述. -
按照1.3中的方法将400 μg脂肪体样品酶解成肽段,按照高pH反相色谱柱(Thermo Fisher Scientific,84868)说明书对肽段进行分级,之后采用上述摸索好的各项条件进行质谱检测后采用Streamine库进行数据匹配.
1.1. 材料
1.2. 液氮研磨与组织研磨器研磨的比较
1.3. 质谱前处理步骤
1.4. 质谱检测
1.5. 质谱数据匹配
1.6. 不同上样时间与不同上样体积对质谱鉴定蛋白质数量的影响
1.7. 不同提取试剂对质谱鉴定蛋白质数量的影响
1.8. 家蚕数据库的优化
1.9. 反相色谱柱分级
-
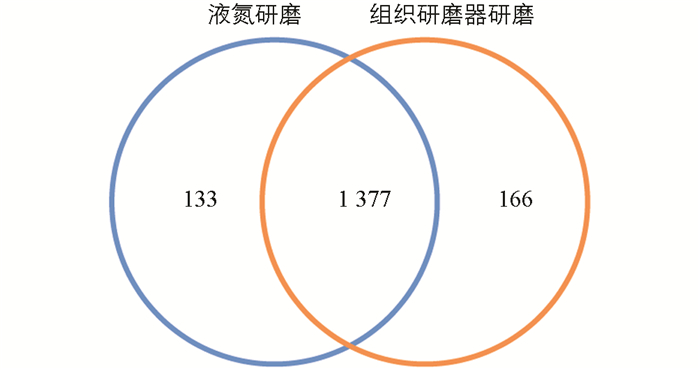
通过比较液氮提取与组织研磨器提取鉴定到的蛋白质数量(图 1),可以发现2种研磨方式鉴定到约90%的蛋白质都一样(1 377种),仅有小部分不同. 组织研磨器提取(166种)比传统液氮提取(133种)鉴定到的蛋白质个数多33种.
-
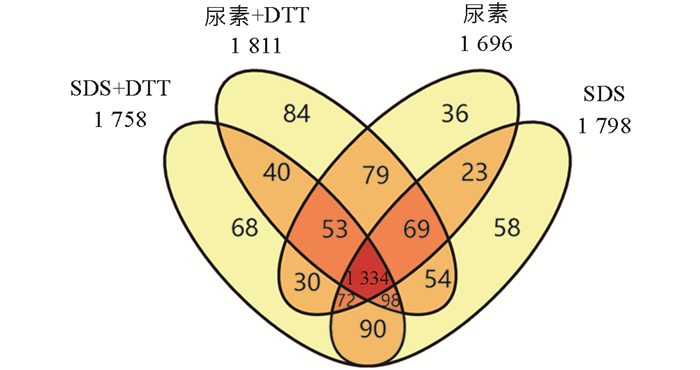
通过对4种常见的蛋白质提取溶液进行比较可以看出(图 2),尿素+DTT溶液鉴定到的蛋白质数量最多,共鉴定到1 811种蛋白质. 尿素鉴定到的蛋白质数量最少,为1 696种. 通过该实验可以看出,SDS溶液和尿素溶液的提取方法都能鉴定到绝大多数相同的蛋白质(1 334种),不同的提取试剂又能提取到一些特有的蛋白质. 通过不同试剂间的比较,最多有115种蛋白质的提升.
-
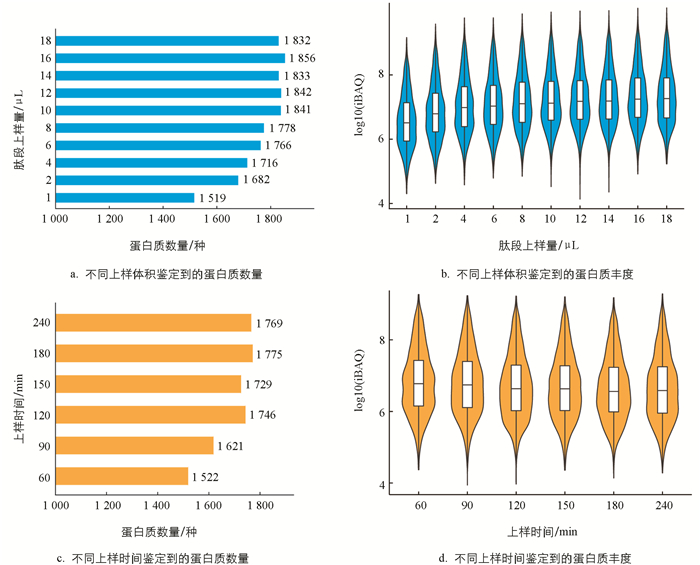
通过比较不同上样体积鉴定到的蛋白质数量,可以看出当上样体积达到10 μL时,鉴定到的蛋白质数量不再明显增加(图 3a). 肽段上样量在4 μL以上时,蛋白质的中位数丰度(iBAQ)趋于稳定(图 3b),蛋白质丰度跟检测响应的信号值相关,因此10 μL的上样量是最佳上样量. 对于家蚕样品,最佳上样时间为120 min(图 3c),而此时蛋白度的丰度值也相差不大(图 3d). 通过不同上样体积的比较,可以提升322种蛋白质. 通过不同上样时间的比较,可以提升224种蛋白质.
-
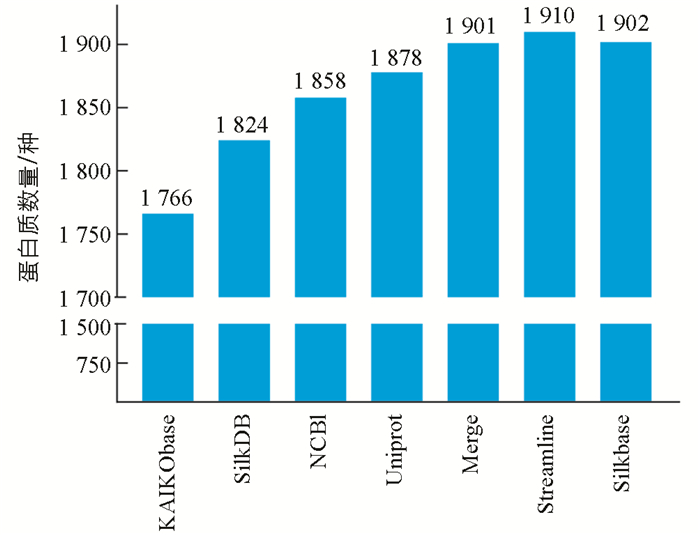
通过用家蚕二代数据库KAIObase,SilkDB,NCBI,Uniprot搜同样的一组家蚕脂肪体质谱原始数据,分别鉴定到1 766,1 824,1 858,1 878种蛋白. 4个家蚕二代数据库的鉴定效果差别不大,平均鉴定数量为1 832种,将4个库整合后的Merge数据库鉴定到1 901种,非冗余数据库Streamline鉴定到1 910种(图 4).
-
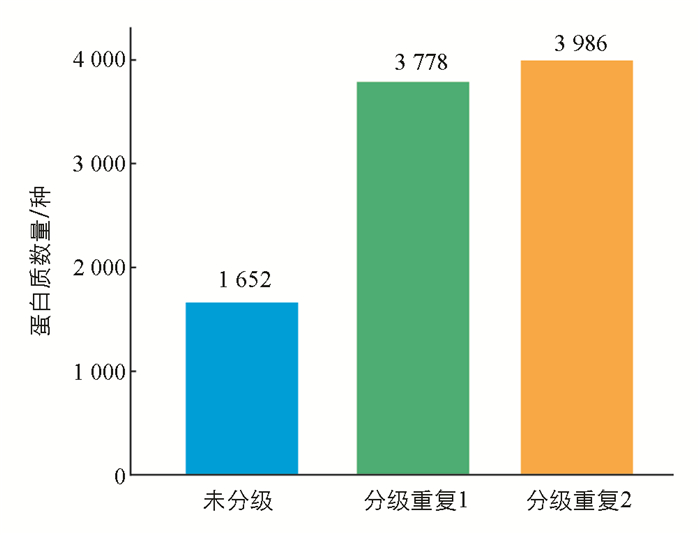
采用反相色谱柱分级肽段的方法对酶解肽段进行分级后,经质谱检测,家蚕脂肪体最多可以鉴定到3 986种蛋白质(图 5).
2.1. 液氮研磨与组织研磨器研磨的比较
2.2. 不同提取试剂对质谱鉴定蛋白质数量的影响
2.3. 不同上样体积和不同上样时间对蛋白质数量的影响
2.4. 家蚕数据库的比较与优化
2.5. 反相色谱柱分级肽段提升蛋白质数量
-
本研究通过对研磨方法、提取试剂、上样体积、上样时间、数据库、分级方法等条件进行实验,发现很多条件都能影响蛋白质的鉴定数量.
比较液氮研磨和组织研磨器研磨,发现两种研磨方式鉴定到的蛋白质数量接近. 在一些组织或器官比较小的样品可以使用组织研磨器提取,该种方法能有效避免蛋白在提取过程中的损失,让质谱鉴定到更多的蛋白质. 液氮研磨需要对研钵和研棒进行清洗、高温消毒等,在进行研磨时需要使用大量的液氮进行降温,不仅浪费液氮,而且还有可能造成样品污染等问题. 组织研磨器研磨样品只需在离心管中进行,研磨样品也只需5 min左右. 综上所述,组织研磨器研磨样品,具有稳定和快速等优点.
样品提取方式多种多样,蛋白质种类繁多,不同的试剂提取蛋白质的效率也不一样,目前没有一种能提取所有蛋白质并用于质谱检测的溶液. 尿素和SDS都能使蛋白质变性而溶解在溶液中,但是哪种溶液更好目前还没有一个准确的依据. DTT加入可还原二硫键,能使蛋白质更好地溶解在溶液中. 通过比较不同的提取溶液,可以看出尿素+DTT这种组合方法鉴定的蛋白质数量最多. 在不加DTT的情况下,SDS的提取效果要优于尿素,而在加DTT的情况下,尿素的效果要优于SDS. 在加入DTT后,无论是尿素还是SDS溶液,鉴定的蛋白质数量都比不加DTT多. 通过提取溶液的摸索,使质谱鉴定数量增加了115种.
通过同一样品不同上样体积的摸索实验可以发现,质谱上样体积达到饱和时,即使再增加上样体积,也不会显著影响鉴定的蛋白质数量. 本研究确定了家蚕样品最佳上样量为10 μL,从所鉴定蛋白质的丰度来看,在10 μL检测的信号响应值也达到最大,通过上样体积的摸索能提升322种蛋白质. 通过对同一样品上样时间的探索,随着上样时间的逐渐增加,在120 min之后鉴定到的蛋白质数量几乎没有提升,因此也可确定120 min为检测的最佳上样时间,该种方法能多鉴定到224种蛋白质. 目前家蚕样品多采用3 h上样时间[29],本方法每个样品能节约1 h检测时间.
在对不同数据库的比较中可以看出,家蚕数据库对鉴定蛋白质数量影响不大,家蚕二代基因组数据库平均鉴定数量为1 832种. 如果简单地将库进行合并,搜库结果中会存在许多冗余数据,非常不利于结果的详细分析. 构建的非冗余数据库Strealine,最多能鉴定到1 910种蛋白质. 值得一提的是,目前日本已经利用第三代测序技术,重新构建了第三代家蚕基因组数据库Silkbase[6],该库中有16 880种蛋白质序列,共匹配到了1 902种蛋白质,彰显出三代数据库的一定优势.
在利用肽段分级策略对样品分级后,可以显著提升鉴定的蛋白质数量,最多鉴定到了3 986种蛋白质. 目前的家蚕脂肪体样品中,很少有文献能达到这种鉴定效果[14, 29-34],如果将来做家蚕大规模蛋白质组测序,使用该种分级策略能大大提高蛋白质的鉴定数量.
-
本研究通过优化蛋白质组质谱前处理中的样品研磨方式和样品提取溶液、优化质谱检测的上样量和检测上样时间、优化数据库和使用分级策略等参数,使家蚕脂肪体鉴定到的蛋白质数量最终提升至3 986种. 解决了家蚕样品量少不易于蛋白质组检测、家蚕蛋白质组最佳上样时间、家蚕数据库冗余等问题,并通过肽段分级策略大大提升了蛋白质的鉴定数量. 建立了较为完善的家蚕蛋白质组检测平台,为大规模蛋白质组测序奠定了基础.
 DownLoad:
DownLoad: