




-
子宫内膜癌(EC)是世界上最常见的女性恶性肿瘤之一[1-4]. 因此,研究EC的分子发病机制,寻找与EC发生、发展和预后相关的生物标志物尤为重要.
长非编码RNA(lncRNA)被定义为长度大于200 bp且无蛋白质编码潜力的转录物[5]. 近年来,越来越多的研究表明,lncRNA在细胞发育、分化、增殖、迁移和转移的生理和病理过程中起着重要作用[6-11]. 研究表明,某些lncRNA可作为潜在的癌症诊断生物标志物[12-14];MALAT1可作为筛查肺癌、子宫内膜间质肉瘤、宫颈癌和肝癌的生物标志物[15]. TRIB3已被证明是EC的潜在治疗靶点,因为它可以通过调节AKT信号通路促进EC细胞凋亡和抑制EC细胞增殖和迁移[16].
内源竞争RNA(ceRNA)假说揭示了RNA相互作用的新机制,认为信使RNA(mRNA)和其他非编码RNA可以通过共同的microRNA(miRNA)反应元件竞争性地与miRNA结合,从而调节某些基因的表达水平[17]. 近年来,越来越多的研究证实ceRNA调控理论与肿瘤的发生、发展和预后密切相关[18].
目前,EC中的ceRNA调节网络机制尚不清楚.
本文先对mRNA,lncRNA和miRNA分别进行差异表达分析;随后通过GO功能富集分析和KEGG通路分析,进一步挖掘了差异表达的mRNA潜在的生物学功能;接着通过成对预测,整合差异表达的mRNA(DEmRNA)、差异表达的lncRNA(DElncRNA)和差异表达的miRNA(DEmiRNA),构建了与EC相关的ceRNA网络,帮助我们挖掘EC发生的分子机制. 为了确定与EC相关的预后因素,对ceRNA网络中的RNA进行了生存分析. 通过多变量Cox回归,构建了一个风险评分系统,对EC患者生存期具有良好的鉴别和预测能力. 本研究对理解EC的分子相互作用机制提供了新的见解,进一步缩小了靶向lncRNA的范围,也将有助于EC的早期诊断、预后及新治疗策略的制定.
HTML
-
本文所使用的数据集来自从肿瘤基因图谱(TCGA)数据库、EC的mRNA(包括lncRNA)和miRNA表达数据及相应临床数据(https://genome-cancer.ucsc.edu/,2019年7月31日更新). 下载的mRNA和miRNA表达数据分别包含583个样本(35个正常样本,548个肿瘤样本)和575个样本(33个正常样本,542个肿瘤样本). 我们用GENCODE数据库(https://www.gencodegenes.org/,版本32)以识别mRNA和lncRNA. RNA表达数据包含19 668个mRNA、14 090个lncRNA和1881个miRNA.
-
应用R软件中的edge软件包筛选548例肿瘤组织与35例正常组织的差异表达的mRNA和lncRNA. 用R软件中的limma软件包对542例肿瘤组织和33例正常组织样本进行了miRNA的差异表达分析[18]. 分别在两组水平分析显著异常表达的lncRNA,miRNA和mRNA:中分化至高分化(G1-G2期)EC样本与正常样本、低分化(G3-G4期)EC样本与正常样本. DEmRNA,DElncRNA和DEmiRNA的筛选标准为:假发现率(FDR) < 0.01且|log2(FC)(fold change)|>2. 然后,用火山图显示符合标准的差异表达的lncRNA,miRNA和mRNA. 另外,通过韦恩(Venn)图显示了G1-G2期与G3-G4期两组样本中相交的异常表达基因,便于进行下游分析.
-
为了探索DEmRNA的潜在生物学功能,利用Database for Annotation,Visualization and Integrated Discovery(DAVID)(https://david.ncifcrf.gov/)数据库对异常表达的基因进行GO功能富集分析和KEGG通路富集分析[19-20]. 在GO和KEGG通路分析中,P.value < 0.01被认为具有统计学意义.
-
为了进一步理解mRNA,lncRNA和miRNA在EC中的相互作用机制,构建了基于DEmRNA,DEmiRNA和DElncRNA相互作用的ceRNA网络. DEmiRNA的靶向lncRNA是基于miRcode数据库[21](http://www.mircode.org/)进行预测. 然后,利用miRTarBase[22](http://mirtarbase.mbc.nctu.edu.tw/),miRDB[23-24](http://www.mirdb.org/)和TargetScan[25](http://www.targetscan.org/)数据库预测miRNA靶向的mRNA. 为了获得更可靠的miRNA与mRNA的关系,利用3个数据库预测结果的交集,建立了一个lncRNA-miRNA-mRNA调控网络. 最后用Cytoscape[26](http://www.cytoscape.org/)软件可视化ceRNA网络.
-
用R软件中的survival软件包对ceRNA网络包含的mRNA,lncRNA和miRNA进行生存分析. 以RNA表达水平的中位数作为截止值,将患者分为高表达组和低表达组. 对数秩P.value < 0.05被认为具有统计学意义. 通过对ceRNA中的RNA进行Kaplan-Meier (K-M) 生存分析,获得了与总体生存时间相关的mRNA,lncRNA和miRNA. 另外,用R软件中的survminer软件包绘制K-M生存曲线,进一步验证mRNA,lncRNA和miRNA的预后价值.
-
构建lncRNA风险评分系统为子宫内膜癌患者的早期诊断提供便利. 将EC患者样本按照1:1的比例随机分为训练集和测试集,然后,基于训练集利用最大似然法建立与生存相关的DElncRNA的Cox风险比例回归模型,并计算模型的回归系数(β). 最后,构建了一个包含5个lncRNA的子宫内膜癌预后风险评分系统.
其中:P表示预后指数(Prognostic index),xi(i=1,2,3,4,5)分别代表WT1-AS,PRICKLE2-AS2,LINC00491,ALDH1L1-AS2和ADAMTS9-AS1的表达水平. 为了评估风险评分系统的识别和预测能力,构建了K-M生存曲线和时间依赖性受试者操作特征(ROC)曲线.
-
为了探索EC患者的临床特征,包括年龄(Age)、临床分期(Clinical stage)、组织学分级(Neoplasm histologic grade)、体重(Weight)和种族(Race)是否与总体生存率有显著相关,我们进行了单变量Cox回归分析. 然后,年龄、临床分期、组织学分级和风险评分水平作为候选变量被纳入多元Cox回归分析. P.value<0.05被认为具有显著统计学意义,并计算各变量的风险比和95%置信区间.
1.1. 数据来源及处理
1.2. 差异分析
1.3. DEmRNA的功能富集分析
1.4. 构建ceRNA调控网络
1.5. 生存分析
1.6. 构建风险评分系统
1.7. 单变量和多变量Cox回归分析
-
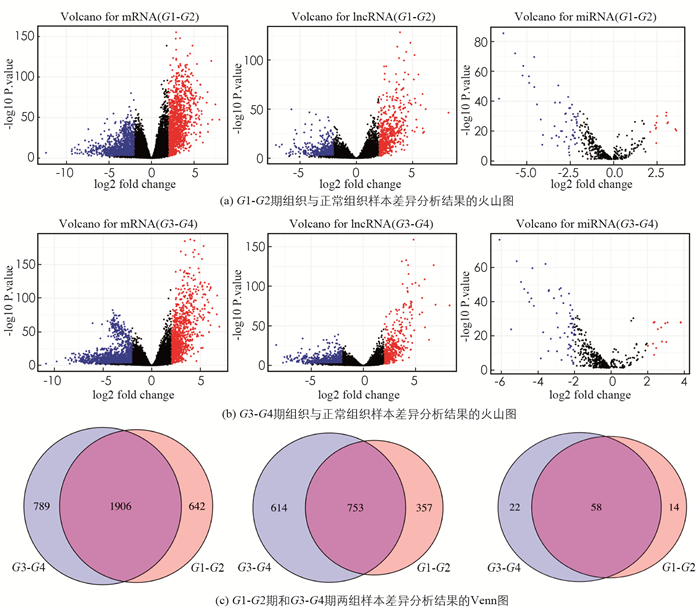
如图 1(a)所示,在G1-G2期的EC组织和正常组织样本中识别了2 548个DEmRNA(1 224个上调,1 324个下调),1 146个DElncRNA(640个上调,506个下调),72个DEmiRNA(13个上调,59个下调);如图 1(b)所示,在G3-G4期的EC组织和正常组织样本中发现了2 695个DEmRNA(940个上调,1 755个下调),1 347个DElncRNA(373个上调,974个下调),80个DEmiRNA(14个上调,66个下调). 两组差异基因的交叉部分由753个lncRNA,58个miRNA和1 906个mRNA组成,这些被认为是早期EC发展的关键基因(图 1(c)).
-
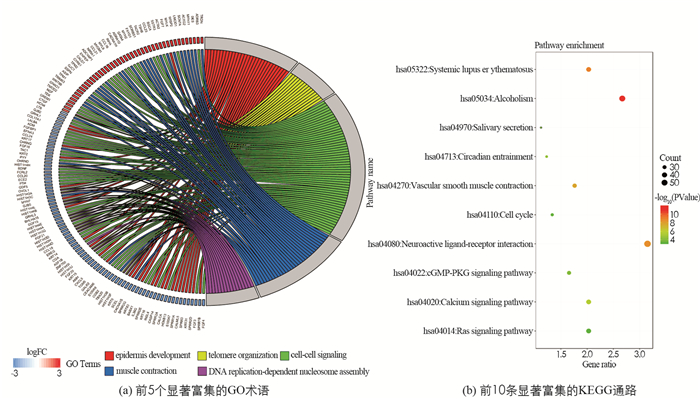
我们进一步研探究了1 960个DEmRNA的潜在生物学功能. 通过GO功能富集分析和KEGG通路分析,筛选出121个显著富集的GO术语(P.value<0.01). 在这些术语中,“表皮发育”“端粒组织”“细胞信号”“肌肉收缩”和“依赖DNA复制的核小体组装”是前5位的GO术语(图 2(a));确定了56条DEmRNA显著富集的KEGG通路、22条KEGG通路在P.value < 0.01时被确定为具有统计学意义,DEmRNA在“hsa04080:神经活性配体-受体相互作用”“hsa04270:血管平滑肌收缩”“hsa04020:钙信号通路”“hsa04022:cGMP-PKG信号通路”“hsa04110:细胞周期”“hsa04014:Ras信号通路”等信号通路显著富集(图 2(b)).
-
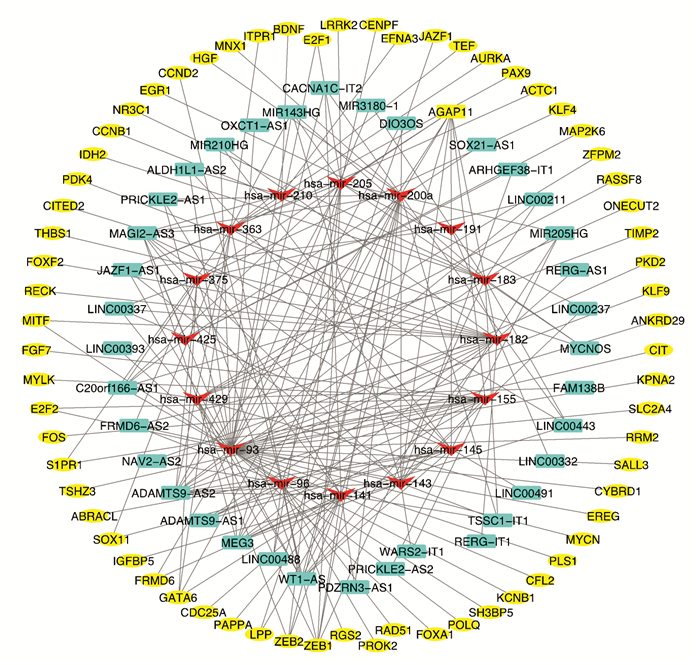
利用miRcode数据集预测753个DElncRNA和53个DEmiRNA,成功地鉴定出136个miRNA-lncRNA对. 然后,利用TargetScan,miRDB和miRTarBase数据库,分析了58个DEmiRNA和1 906个DEmRNA,发现了84个miRNA和mRNA相互作用对. 最后构建了一个包含66个mRNA,16个miRNA和37个lncRNA的ceRNA调控网络(图 3).
-
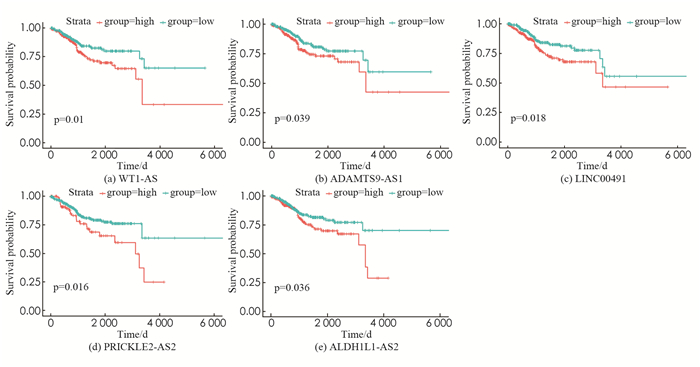
为了确定与EC患者预后相关的mRNA,lncRNA和miRNA,对ceRNA中的每个RNA(66个mRNA,37个lncRNA和16个miRNA)进行了K-M生存分析和Log-Rank检验. 最后发现,16个mRNA(NR3C1,CIT,SOX11,CDC25A,RECK,AURKA,E2F1,ONECUT2,SALL3,SLC2A4,GFBP5,POLQ,RGS2,MNX1,KLF9和RRM2),5个lncRNA(WT1-AS,PRICKLE2-AS2,ADAMTS9-AS1,ALDH1L1-AS2和LINC00491)和1个miRNA(hsa-mir-182)与EC患者的总体生存率显著相关(P.value < 0.05)(图 4).
-
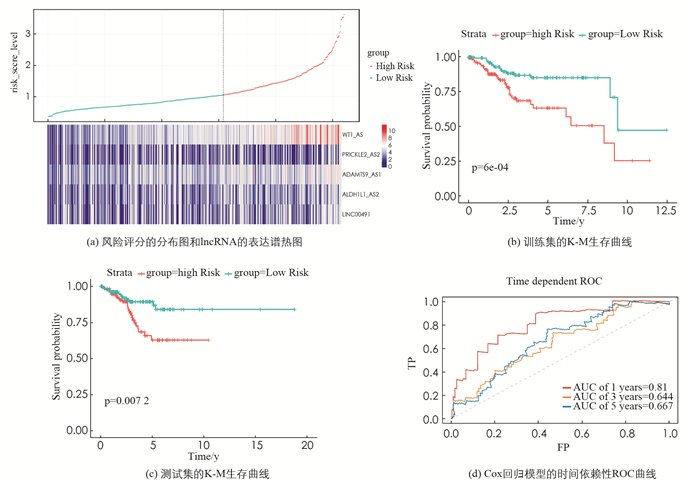
基于与总体生存率显著相关的5个lncRNA,应用多元Cox回归分析来构建风险评分系统,其贡献由其相关系数加权,最终的风险评分公式为:
其中:P表示预后指数(Prognostic index),xi(i=1,2,3,4,5)分别代表WT1-AS,PRICKLE2-AS2,LINC00491,ALDH1L1- AS2和ADAMTS9-AS1的表达水平. 风险评分大于最佳截断值0.945的患者被视为高危患者(212名患者),而风险评分小于或等于0.945的患者被视为低危患者(308名患者). 特别的,根据K-M和时间依赖性ROC曲线分析,这两个组的设计均提高了对子宫内膜癌高、低危患者的预测正确率(图 5(b),(c)). 基因表达热图和患者评分散点图(图 5(a))显示了520例EC患者生存期的5个lncRNA表达谱和风险评分以及垂直虚线0.945的风险评分的截止值. 采用单因素Cox回归分析筛选520例临床资料完整的EC患者总体生存率相关的特征,结果表明,年龄、临床分期、组织学分级的预后价值具有统计学意义. 在多因素Cox回归分析中,年龄、临床分期、组织学分级和风险评分与EC患者的预后密切相关. 因此,我们构建的lncRNA风险评分系统可以作为子宫内膜癌患者的独立预后指标(表 1).
2.1. 差异表达分析
2.2. DEmRNA的功能富集分析
2.3. 构建ceRNA调控网络
2.4. 生存分析
2.5. 构建与lncRNA相关的风险评分系统
-
子宫内膜癌是一种致命的女性恶性肿瘤. 在过去的20年里,EC死亡率翻了一番. 只有20%的EC患者在绝经前被诊断出来[27-28]. 因此,研究EC的分子发病机制,寻找与EC发生、发展和预后相关的生物标志物尤为重要. 我们首先对收集到的EC患者的样本,分别对mRNA,lncRNA和miRNA数据进行了差异表达分析,最终得到在EC组织中异常表达的mRNA,lncRNA和miRNA. 然后,在此基础上通过成对预测获得了miRNA的靶向lncRNA和mRNA. 最终构建了EC的一个ceRNA调控网络进一步去理解EC分子间相互作用机制. 接下来,通过对包含在ceRNA网络中的RNA进行了生存分析,筛选出了与EC患者总体生存显著相关的mRNA,lncRNA和miRNA. 最后,根据筛选出的5个与EC患者总体生存时间密切相关的lncRNA,利用Cox多元回归构建了一个风险评分系统. K-M生存曲线和时间依赖性ROC曲线进一步验证了该风险评分系统具备良好的预测能力,有助于子宫内膜癌早期诊断. 单因素Cox回归和多因素Cox回归分析的结果也证明了这个风险评分可以作为EC患者生存时间的独立预后指标. 本研究深入挖掘了EC的分子相互作用机制并进一步缩小了靶向lncRNA的范围,将有助于EC的早期诊断、预后及新的治疗策略的制定. 由于缺乏其它类似的EC相关lncRNA数据库,因此未进行外部验证.
 DownLoad:
DownLoad: