


 下载:
下载:
-
番茄是世界范围内广泛种植的蔬菜作物之一. 同时, 我国是种植面积最大和产量最高的番茄种植国家(FAO数据). 然而, 在番茄种植和生长发育过程中, 各种生物和非生物胁迫常常严重制约了其生长发育, 从而影响了蕃茄的产量和品质. 随着遗传转化技术和分子生物学技术的发展, 番茄也逐渐发展成为一种模式研究作物[1], 这极大促进了人们对茄科作物的信号转导通路的认识. 已有的研究表明, 植物通过一系列的信号转导过程来准确应答和抵御外界的不利因素[2-3]. 例如, 在病原菌入侵过程中, 植物就存在病原模式相关分子诱导的免疫(PTI)和效应蛋白诱导的免疫(ETI)两种不同形式的应答机制[4]. 有趣的是, 植物的各个信号通路之间存在着复杂的交互关系(拮抗或者协同促进), 比如, 在番茄的研究中已证实响应病菌侵染和盐胁迫的信号转导通路也存在着交联[5]. 在信号转导过程中, 蛋白之间的相互作用十分关键[6]. 因此, 互作蛋白的鉴定成了研究番茄细胞内响应外界胁迫的信号通路的关键.
酵母双杂交系统是广泛使用的筛选和检测蛋白质相互作用的方法之一[6]. 该技术首次由Fields等[7]提出, 利用酵母菌中GAL4转录因子的DNA结合结构域(binding domain, BD)和转录激活结构域(active domain, AD)在空间上稳定接近时能起到重现完整GAL4转录因子的特点, 来检测分别融合表达BD和AD的两个蛋白是否互作. 酵母双杂交技术已经成功应用于多种蔬菜作物的蛋白互作研究, 例如: 抗番茄叶霉病免疫应答的番茄酵母双杂交cDNA文库[8], 响应干旱胁迫的Micro-TOM番茄酵母双杂交cDNA文库[9].
为了深入研究番茄响应生物胁迫和非生物胁迫时的分子机制, 本研究以“HeinZ1706”番茄为材料, 分别收集其在TMV和辣椒疫霉菌(Phytophthora capsici)侵染下和干旱胁迫下的叶片和根部组织, 采用重组连接的方法, 构建了番茄酵母双杂交cDNA文库, 并通过文库滴度测定、重组率和插入片段大小进行了文库质量评估. 最后, 以在细胞核质定位的辣椒疫霉菌RxLR效应蛋白PcAvr3a11为诱饵蛋白, 利用本次构建的番茄酵母双杂交cDNA文库进行文库筛选, 获得了25个PcAvr3a11的候选互作蛋白. 这些结果为进一步研究PcAvr3a11影响寄主植物免疫的机制奠定了一定基础, 也进一步表明该文库的建立对开展病原菌与番茄互作机制和番茄响应干旱胁迫的机制研究可以起到积极的促进作用.
全文HTML
-
“HeinZ1706”番茄在22~25 ℃, 16 h光照与8 h黑暗交替的光照培养室培养. TMV毒株由西南大学植物保护学院孙现超课题组保存, 采集被TMV完全侵染的本氏烟叶片进行摩擦接种. 辣椒疫霉菌菌株BS11在25 ℃黑暗、10%V8固体培养基上进行培养, 利用获得的游动孢子进行接种.
将苗龄为5周的“HeinZ1706”分为3组(每组2棵), 分别进行摩擦接种[10]野生型TMV病毒[11], 离体叶片接种辣椒疫霉菌游动孢子和干旱胁迫处理. TMV病毒侵染组出现卷叶、斑点等病毒症状时收集番茄叶片组织;接种辣椒疫霉菌游动孢子后, 分别在6, 12, 24 h收集番茄离体叶片组织. 干旱胁迫是进行缺水3 d, 叶片出现萎焉时收集番茄叶片和根部组织.
-
采用Trizol法, 将3种不同处理的番茄组织等量混合提取番茄总RNA, 利用Oligotex mRNA Kits(Qiagen)分离纯化样本的mRNA, 用1%的琼脂糖凝胶和NanoDrop2000分光光度计检测其质量和浓度. 获得的mRNA条带清晰, 呈弥散状分布, 条带分布均匀, 总量大于4.5 μg为合格.
-
RT-preimer(5’-TCATCTGCAGCTCGAGCACAACTTTGTACAAGAAAGTTGGGTTTTTTTTTT TTTTTTTTTTTVN-3’)为引物, 取4.5 μg mRNA为模板, 利用SuperScriptTM IV Reverse Transcriptase (Thermo Scientific)合成第1链cDNA. 以第1链cDNA为模板, 利用Second Strand cDNA Synthesis Kit (Thermo Scientific)合成cDNA第2条链. 以获得的双链cDNA为模板, 利用T4 DNA ligase (NEB)加入接头. 3个读码框, 每个读码框1份. 接头序列为(5’-CAGATTACGCTCATATGACAACTTTGTACAAAAAAGTTGG(AA)-3’). 最后加入2 μL 10 mmol/L dNTP, 2 μL T4 DNA聚合酶, 16 ℃放置20 min, 补平末端.
-
将得到的cDNA分别进行乙醇沉淀, 最终溶解在14 μL DEPC水中, 取12 μL进行后续反应, 加入4 μL 4X杂交buffer, 98 ℃ 2 min, 68 ℃ 5 h. 将反应管保持在68 ℃, 加入4 μL 5X DSN buffer, 然后加入0.2 μL的DSN酶(1 U/μL), 置于68 ℃反应3 min. 加入10 μL EDTA, 混匀, 加入等体积的酚氯仿抽提一次. 将抽提产物进行乙醇沉淀, 溶于80 μL DEPC水中. 取1 μL保存. 以剩余79 μL cDNA为模板, 以5’-CAGATTACGCTCATATGACA-3’和5’-TCATCTGCAGCTCGAGCACA-3’为引物, 利用DNA聚合酶反应扩增5个循环. 用1%质量浓度的低熔点琼脂糖胶电泳检测cDNA产物, 切胶回收500 bp以上的片段, 获得均一化处理的cDNA.
-
将7 μL的cDNA与3 μL的经酶切(NdeⅠ/XhoⅠ)处理线性化的pGADT7载体混合, 加入5 μL的Infusion重组酶[12], 以及5 μL的DEPC水, 混匀, 置于50 ℃反应1 h. 通过电击转化高效率的大肠杆菌感受态细胞DH5α, 将所有重组反应的产物转化到大肠杆菌中. 将转化后的大肠杆菌DH5α置于37 ℃, 225~250 r/min培养1 h. 培养结束后, 吸取10 μL培养物稀释10, 100, 1 000, 10 000倍, 分别取10 μL稀释液涂布LB平板(含50 mg/L氨苄青霉素), 37 ℃培养过夜, 第2 d计数菌落数, 获得文库初始滴度. 随机挑取24个克隆, 以pGADT7-F和pGADT7-R为引物, PCR扩增, 电泳检测重组率和插入片段平均大小. 剩余培养物, 每10 μL涂布一个LB平板, 37 ℃培养过夜, 第2 d用涂布器刮取所有菌落混合. 取1 mL菌加入甘油(甘油最终浓度为20%)后液氮速冻保存到-80 ℃冰箱;其他菌用于质粒提取, 获得酵母双杂交cDNA文库筛选的质粒.
-
以辣椒疫霉菌LT1534 cDNA为模板, 设计特异性引物PcAvr3a11-F和PcAvr3a11-R扩增编码PcAvr3a11成熟区段的基因序列(去掉了分泌信号肽). 通过限制性内切酶的酶切、胶回收、T4 DNA ligase连接和测序验证得到pGBKT7: : PcAvr3a11和pART27: : GFP-PcAvr3a11载体. 将pART27: : GFP-PcAvr3a11载体转入农杆菌GV3101中, 利用根癌农杆菌介导的本氏烟瞬时表达技术[13]和激光共聚焦观察, 明确PcAvr3a11在植物细胞内的定位情况. 利用PEG/LiAc介导的酵母转化方法, 将诱饵载体pGBKT7: : PcAvr3a11和pGADT7空载体共转入酵母菌株AH109, 在缺陷型培养基上检测PcAvr3a11是否具有自激活.
-
大规模的酵母双杂交文库筛选参照YeastmakerTM Yeast Transformation System 2试剂盒(Clontech) 说明书进行. 将转子涂布在SD/-Leu-Trp-His-Ade培养基上, 4~5 d后再将四缺培养基上生长出来的酵母菌落挑取到加有X-α-Gal的SD/-Leu-Trp-His-Ade培养基上, 挑取生长并变蓝的酵母单菌落, 利用玻璃珠破壁法提取酵母质粒. 将酵母质粒转化大肠杆菌后送测序, 获得候选互作蛋白的基因序列. 将基因序列比对到番茄基因组数据库, 获得候选互作蛋白的注释情况.
1.1. 实验材料和处理方法
1.2. 总RNA的提取和mRNA的分离
1.3. 双链cDNA合成
1.4. DNA的均一化处理
1.5. cDNA与核蛋白酵母双杂交载体pGADT7的重组和文库滴度鉴定
1.6. 诱饵载体构建、亚细胞定位和酵母自激活鉴定
1.7. 酵母双杂交文库筛选
-
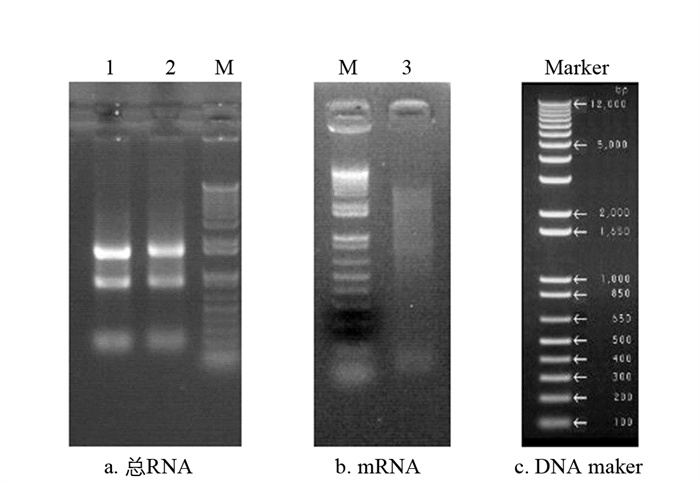
按照1.1的方法, 将3种处理收集的番茄组织等质量混合, 利用Trizol试剂提取番茄总RNA. 在1%琼脂糖胶中检测总RNA的提取质量. 如图 1a所示, 28 S和18 S两条带清晰明亮, 无明显弥散性条带. 由此判断番茄cDNA文库的总RNA质量较好, 无明显降解. 利用Oligotex mRNA Kits(Qiagen)分离纯化获得番茄样本的mRNA. 如图 1b所示, mRNA条带在500~3 000 bp范围内呈均匀弥散分布, 说明cDNA文库mRNA质量较好, 满足文库构建要求.
-
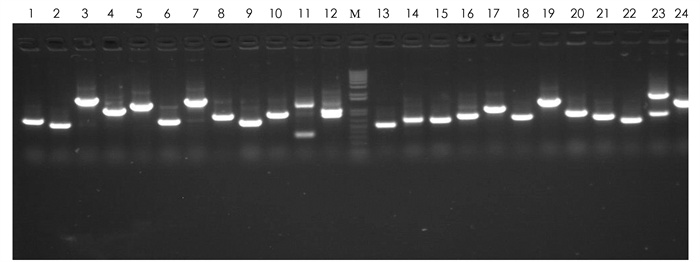
以获得的mRNA为模板, 经均一化处理最终获得ds cDNA, ds cDNA重组连接到pGADT7载体后, 通过电击转化高效率的大肠杆菌获得酵母双杂交cDNA文库. 吸取10 μL初始培养物, 稀释10, 100, 1 000, 10 000倍, 分别取10 μL稀释液涂布LB平板将其涂布到平板上, 计算得到初始文库滴度约1.15×107CFU. 在cDNA文库中随机挑取24个单菌落, 进行PCR和琼脂糖凝胶电泳检测. 如图 2所示, 24个克隆全部扩增出条带, 重组率为100%, 条带主要分布在500~2 000 bp, 平均长度约为1 kb, 条带具有较好多态性. 这表明本次番茄酵母双杂交文库构建满足未来实验需求.
-
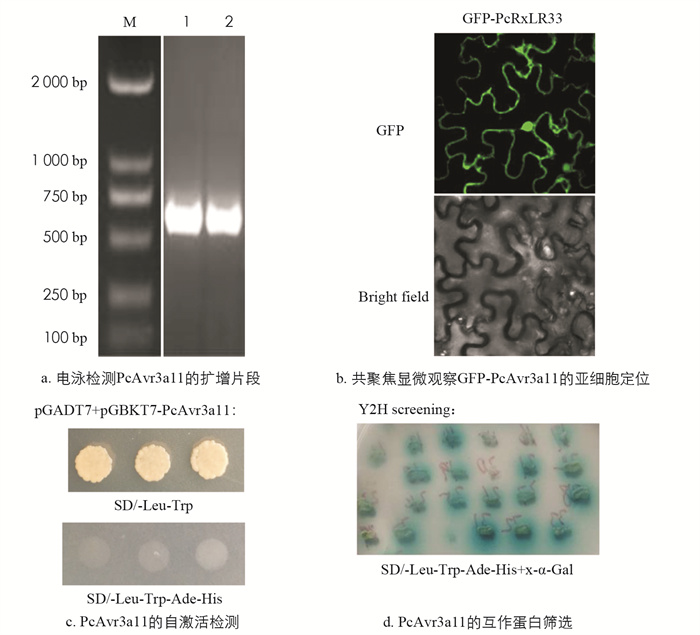
以辣椒疫霉菌cDNA为模板, PCR扩增获得编码辣椒疫霉菌RxLR效应蛋白PcAvr3a11成熟区段的基因序列(图 3a). 通过酶切、连接和测序验证得到pGBKT7: : PcAvr3a11和pART27: : GFP-PcAvr3a11载体. 将pART27: : GFP-PcAvr3a11载体转入农杆菌GV3101中, 利用根癌农杆菌介导的本氏烟瞬时表达技术[11]和激光共聚焦观察明确PcAvr3a11在植物细胞内的亚细胞定位. 如图 3b所示, GFP-PcAvr3a11在细胞核和细胞质内都有. 这符合pGADT7系统酵母双杂交检测细胞核和细胞质蛋白之间互作的要求. 利用PEG/LiAc介导的酵母转化方法, 将诱饵载体pGBKT7: : PcAvr3a11和pGADT7空载体共转入酵母菌株AH109. 共转化菌株在缺陷型培养基SD/-Trp-Leu-Ade-His上的生长情况表明, pGBKT7: : PcAvr3a11不具有自激活(图 3c). 结果均表明, PcAvr3a11适用于酵母双杂交文库筛选的诱饵蛋白.
-
将番茄酵母cDNA文库质粒和pGBKT7: : PcAvr3a11共转化入酵母感受态细胞Y2H Gold. 取10 μL转化酵母菌液稀释1 000倍后涂布1 00 μL至SD/-Trp-Leu平板培养基上, 计算该次筛库的总转化子数目约为5×105, 理论上已完全覆盖番茄全基因组, 满足酵母双杂交文库筛选的要求. 将剩余未稀释菌液涂布于50个SD/-Leu-Trp-Ade-His培养皿上进行筛选, 再将生长的阳性克隆挑到SD/-Leu-Trp-Ade-His + X-α-Gal培养皿上(图 3d), 共获得33个转变为蓝色的阳性生长菌落. 测序结果经过序列相似性对比和重复序列分析后得到25个可能与PcAvr3a11互作的候选基因(表 1).
2.1. 番茄总RNA和mRNA的质量检测
2.2. 酵母双杂交cDNA文库构建和质量评估
2.3. 诱饵蛋白酵母双杂交载体的构建及其自激活检测
2.4. PcAvr3a11互作蛋白的初步筛选及其序列分析
-
酵母双杂交技术是筛选互作蛋白的常用手段之一[6], 对研究植物响应外界胁迫过程中的蛋白作用机理、信号转导过程与代谢途径具有重要帮助. 酵母双杂交cDNA文库构建方法主要有Takara公司推出的SMART法和Invitrogen公司推出的Gateway法. 本研究利用重组酶一步连接到线性化目的载体pGADT7的方法, 既避免了SMART方法中Sfi1酶切cDNA片段时可能导致基因断裂的问题, 也避免了Gateway方法中两步重组反应会放大基因复制偏好性的问题. 本研究在cDNA的5’端做了3份不同读码框的接头, 尽量保证了插入基因的正确表达. 最后, 为了降低高丰度基因的相对含量, 使用DSN法对其进行均一化处理, 降低高丰度基因. 该文库的初始库容量达到了1.15×107 CFU, 重组阳性率为100%, 平均插入片段大小约为1 kb, 理论上达到了酵母双杂交cDNA文库构建的要求.
PcAvr3a11是来自辣椒疫霉菌的典型RxLR效应蛋白之一[14]. 本研究实验表明PcAvr3a11在酵母系统中不具有自激活且定位于植物细胞核和细胞质中. 因此, 酵母双杂交文库筛选适用于寻找PcAvr3a11在寄主植物中的互作蛋白. 利用已构建的酵母双杂交文库, 以PcAvr3a11为诱饵蛋白进行了酵母双杂交文库筛选, 最终获得了25个候选互作蛋白. 这进一步表明利用重组酶一步连接的方法构建酵母cDNA文库具有可行性. 通过农杆菌侵染法[15], 能较为高效地构建转基因番茄. 未来通过在植物上的进一步验证, 可以确认哪些蛋白是被PcAvr3a11靶向并具有生物学意义的植物蛋白. 这些候选互作蛋白为研究PcAvr3a11如何干扰寄主植物细胞奠定了一定的研究基础.