


 下载:
下载:
-
在未来的20年里人们对动物蛋白的需求将不断增长,而动物蛋白主要来源于牛奶和家禽产品[1],这使得研究者们相信有必要提高畜禽产量,在发展中国家更有必要.饲养蛋鸡的主要成本是饲料,一般可以通过修改饮食配方,从而提高饲料效率的方法来减少用量.随着生物技术的发展,使用基因选择则是一种更有效的方法[2].
肠道既是重要的消化器官,又是维持内环境稳态的天生屏障.肠道发育是家禽饲料效率的一个重要因素,与消化酶活性和吸收营养的能力密切相关.肠道长度是消化道发育的一个重要指标,鸡肠道的长度与采食量有关,并影响鸡的生长[3].盲肠被认为是鸡消化粗纤维的主要场所,其长度与消化能力相关[4].虽然有一些文献报道了小肠长度的遗传背景[5-8],然而很少有关于盲肠长度遗传背景的研究报道.因此,对鸡盲肠长度的遗传结构进行解析很有必要.
将整个基因组层面的序列变异(主要是单核苷酸多态性,SNPs)与表型和血统信息相结合,可用于进行全基因组关联分析(GWAS),并识别感兴趣性状特征的基因或调控元素[9].与传统数量性状基因座(QTL)定位策略相比,GWAS的主要优势在于其检测变异效果的强大能力并将变异定义到较窄的基因组区域[10].在过去的几年中,GWAS揭示了许多与鸡的形态特征、生产性状、抗病等相关的重要研究成果[9].然而,还没有鸡盲肠长度的GWAS研究.因此,本研究主要使用600 K鸡SNP芯片对黑羽蛋鸡F2代资源群体(1 512只母鸡)运用单变量GWAS检测与盲肠长度表型相关联的基因组区域或基因.
全文HTML
-
F2代资源群体由白来航(WL)和东乡绿壳蛋鸡(DX)2个纯系杂交产生. 6只WL公鸡与133只DX母鸡交配、6只DX公鸡与80只WL母鸡交配形成F1代.然后,从WL/DX杂交产生的25只公鸡和407只母鸡与从DX/WL杂交产生的24只公鸡和235只母鸡选留下来生产F2代.共生产3 749只F2代个体(1 856只公鸡和1 893只母鸡).然后,从550个全同胞和49个半同胞家系中选择1 534只母鸡进行SNP基因分型,以确保足够的表型和背景信息.
-
为描述盲肠长度的遗传结构,将72周龄F2代鸡屠宰,收集整个盲肠,挤压出内容物后测量其长度.使用所有可用的记录,运用SAS软件包的MEANS程序对数据进行描述性统计计算.对偏离正态分布的特征,使用SAS软件进行正态转换后再进行关联分析,然后这些转换值被用于下游分析,包括GWAS和遗传估计.
-
F2代鸡进行静脉采血,使用标准的苯酚/氯仿萃取法提取基因组DNA,使用600 K Affymetrix Axiom鸡基因芯片进行基因分型.删除7 883个未知基因位置的SNP和112个冗余基因坐标的SNP,使用Affymetrix工具v1.16.0软件的Axiom GT1算法收集SNP原始数据.只将质量控制(QC)在0.82或更高的样品用于下游分析,共得到1 512个样本和532 299个SNP.此外,考虑到当前统计方法检测表型和常染色体基因型之间的联系更有效,将性染色体的6 402个SNP剔除,使用PLINK v1.90程序包删除67 330个最小等位基因频率(MAF)小于5%和22 700个偏离哈迪温伯格平衡测试(p<1×10-6)的SNP,再使用BEAGLE v4.0程序归因一些零星的缺失基因型,只保留质量分数(R2)大于0.5的SNP[11].最后,共有1 512个样本和435 867个SNP可进行GWAS.
-
GWAS之前先进行主成分分析,消除虚假相关.使用simpleM法更正测试数量,建立全基因组潜在和显著关联的合适阈值.建立有效的独立测试数值Meff=59 308,全基因组显著和潜在的p值分别为8.43×10-7(0.05/59 308)和1.69×10-5(1.00/59 308).首先使用GEMMA v0.94软件的精确混合模型方法进行单变量分析,确定MAF≥0.05的SNP的关联性[12],再用独立SNP计算集中相似度矩阵,然后针对SNP和表型之间的显著水平计算Wald测试的p值.单变量线性混合模型为
y是n个个体的n×1表型值向量,W是n×c协变量矩阵(固定效应),α是c×1对应系数向量,x是n×1标记基因型向量,β是标记的相应效应大小,μ是协方差结构的n×1随机多基因效应向量,ε是n×1随机残差向量.
曼哈顿和quantile-quantile(QQ)图使用R软件附带的GAP包绘制.用于确定假阳性信号程度的基因组膨胀因素λ,也使用R软件附带的GenABEL包中的estlambda函数计算[13].
-
运用GCTA v1.24程序中的单变量限制极大似然法估计遗传力[14].针对各部分肠长度运用二元混合模型估计遗传和表型相关[15].对于全基因组显著SNP,表型方差贡献使用下面的混合线性模型估计为
y是n个个体的n×1表型值向量,b是固定效应向量,X是它的关联矩阵,gG是SNP聚合效应的向量,e是随机残余项.
-
对显著性SNP进行功能注释,使用变异效应预测软件VEP和Ensembl中的Biomart工具,对在给定基因组区域中的候选基因的显著性基因座进行搜索[16-17].
1.1. 构建资源群体
1.2. 表型测量
1.3. 基因分型、质量控制
1.4. 全基因组关联分析
1.5. 遗传和表型方差估计
1.6. 基因鉴定
-
屠宰72周龄F2代鸡,得到盲肠长度CL.共测量1 505只母鸡,获得1 434份有效数据.对其进行描述统计可知,CL平均为(14.94±2.10) mm,最大值、最小值分别为22.30 cm,6.10 cm,变异系数为14.06%.所有表型值在逆正态转换后符合正态分布,转换后的值被用于所有后续分析.
通过合格的GWAS标记对CL及相关性状十二指肠长度DL、空肠长度JL、回肠长度IL的加性遗传方差进行计算,并统计遗传参数,结果如表 1所示.单变量GCTA分析显示CL遗传力在中等水平(h2=0.39).双变量GCTA分析表明,CL与IL的遗传相关较高,而与DL和JL表现出中等遗传相关.表型相关分析表明CL与其他几种小肠长度具有较低的表型相关性.
-
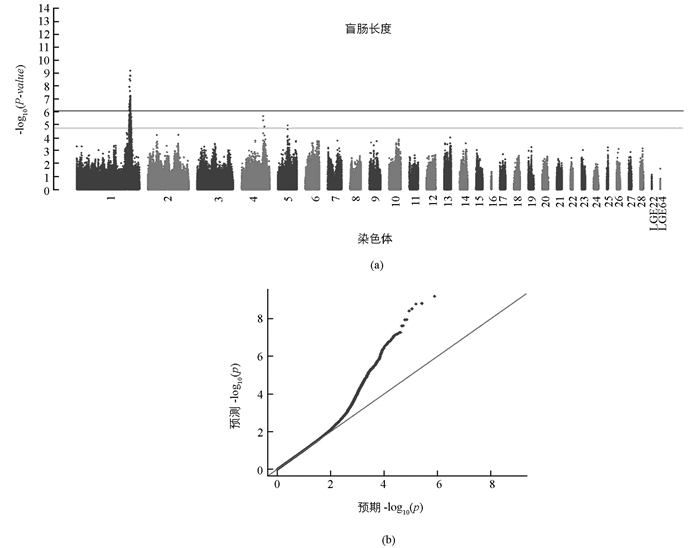
对CL进行单变量全基因组关联分析.共鉴定出54个全基因组显著位点与CL关联.显著的基因组区域从1号染色体上(GGA1)的166.66 Mb到171.06 Mb.分析发现有209个全基因组潜在位点与CL关联,这些位点分别位于GGA1的165.16~172.54 Mb,GGA4的688.54~731.56 Mb以及GGA5的312.90 Mb.根据所有影响盲肠长度的SNP的p值制作曼哈顿图和QQ图,如图 1所示.
-
运用VEP和Biomart对显著SNP周围的区域进行扫描,鉴定与盲肠长度相关的基因.由于基因内部的突变比位于基因间区域的SNP更有意义,我们对上述54个SNP进行了分析.覆盖26个SNP的18个基因被确定为候选基因,其中5个基因有不止1个SNP位点(表 2).在这26个SNP位点中,有2个分别位于CDS和3′UTR,均可被转录成成熟的mRNA.这2个SNP位点rs316180141和rs14914978及其对应的基因NHLRC3和SIAH3被视为最重要的SNP位点和基因.
-
使用GCTA工具对和盲肠长度关联的这2个SNP位点进行表型方差贡献的估计,它们对于盲肠长度的CPV如表 3所示.位点rs316180141和rs14914978分别可以解释CL表型方差的4.11%和3.50%.
2.1. 表型和遗传参数
2.2. 候选位点GWAS鉴定
2.3. 显著位点的基因注释
2.4. 表型方差贡献(CPV)的估计
-
盲肠是显著影响鸡消化吸收粗纤维的重要器官.大部分关于鸡盲肠长度的研究集中在饲料营养方面[18-19],然而对鸡盲肠长度的基因组结构目前尚不清楚. GWAS是分析动物重要遗传结构特点的一个强大工具.目前,在多种农业动物如牛[20-21]、猪[22-23]和家禽[24-25]中已确定了许多QTL及SNP.因此,有必要使用GWAS研究鸡盲肠长度.本研究中包括1 512只蛋鸡,使得特征差异最大化,提高了确定性状QTL的概率,同时高密度(600 K)染色体SNP芯片覆盖了整个鸡染色体,以提高准确性和可靠性.
从盲肠长度的数据可以看出,尽管最大值和最小值之间的差异很大,但大多数测量的数值在正常范围内.遗传评估的结果表明,CL具有中等遗传力.盲肠长度与小肠长度的3个特征相关分析结果表明,其与回肠长度具有高遗传相关性,但表型相关性并不高,表明这些特征受环境因素影响较大.这个结果与我们之前关于小肠长度和质量的研究结果类似.十二指肠、空肠和回肠长度(质量)有较高的遗传相关性,但却是中等表型相关,表明小肠长度和质量也有相似的遗传特征.
GWAS的结果显示,对于盲肠长度大部分的显著性SNP位点位于GGA1的170 Mb附近,表明该区域对盲肠长度很重要.在我们之前对小肠长度和质量的研究中,最重要的SNP位点也落在GGA1的170 Mb附近,表明这个区域对于小肠长度和质量也很重要.由此可见,这段区域是研究肠道发育的重要区域,值得做进一步研究.
在与盲肠长度相关联的26个候选SNP和相对应的18个基因中,我们推测2个SNP位点rs316180141和rs14914978及其对应的基因NHLRC3和SIAH3是最重要的基因,因为这2个SNP位于相应基因的转录区域. NHLRC3(NHL repeat containing 3)是NHL重复包含蛋白3,它可能参与了一些酶过程,比如泛素化等,我们预测它在鸡盲肠中通过各种酶对消化起到促进作用. SIAH3 (siah E3 ubiquitin protein ligase family member 3)是siah E3泛素蛋白连接酶家族成员3,它参与泛素依赖的蛋白水解过程[26],对多器官发育起重要作用.因此,我们认为这2个基因与盲肠长度高度相关,值得进一步研究.而对应的2个SNP的CPV均较高,进一步提示这2个基因是影响鸡盲肠长度的候选基因.
-
通过GWAS鉴定出54个与盲肠长度显著相关的SNP位点,且大部分集中在1号染色体170 Mb附近.共将覆盖26个SNP位点的18个基因定为候选基因,其中2个分别在CDS和3′UTR,对应于NHLRC3和SIAH3.这些位点和基因可能是影响盲肠长度的重要SNP位点和基因.