




-
开放科学(资源服务)标志码(OSID):

-
中药丹参Salvia miltiorrhiza为唇形科鼠尾草属多年生草本植物,其根茎可入药;在中国、日本、美国和欧洲,丹参已广泛用于治疗血管疾病,包括动脉粥样硬化、高血压、高血脂和中风等[1-2]. 近些年的研究表明,丹参还具有其他多种药理活性,如抗氧化、神经保护、抗纤维化、抗炎、抗肿瘤等,其临床应用为患者护理和人体健康做出了重要贡献[3]. 丹参原产于中国,在韩国、越南、澳大利亚等国家也被广泛种植[4]. 国内丹参的主要产区有河南、山东、陕西、山西、湖北、安徽、四川等地[5].
遗传多样性作为生物多样性的核心部分,是长期进化的产物,对丹参药材的利用具有重要意义,也是药用植物适应生存和进化的前提. 植物各种高产、抗病、抗逆等优良性状基因以及控制有效成分代谢途径的基因构成了其基因水平上的多样性,这些遗传多样性为我们收集、保存、利用、评价植物资源提供了依据;因此遗传多样性是中药丹参种质鉴定、资源利用、引种栽培、资源保护的基础[6]. 目前,采用的丹参种源鉴定方法主要有来源鉴定法、性状鉴定法、理化鉴定法和生物鉴定法[7]. 有学者研究了12种丹参的性状、显微特征、理化鉴别特征,并通过化学成分定性定量及药理实验比较分析,评估了丹参种质的质量[8].
DNA标记是研究植物遗传多样性的有力工具[9],主要有叶绿体和核基因标记. 学者们曾采用随机扩增多态性DNA[10-11]、AFLP(扩增片段长度多态性)[12-13]、ISSR(简单重复序列间扩增标记)[14]、叶绿体基因SSR(简单重复序列标记)[15]、EST-SSR(基于表达序列标签开发微卫星标记)[16]对丹参遗传多样性进行了研究. 高等植物细胞核核糖体DNA(Nuclear Ribosomal DNA,nrDNA)是高度重复的串联序列单元,包括1个单元段,1个操纵子,以及由外转录间隔区(External Transcribed Spacer,ETS)、非转录间隔区(Non-Transcribed Spacer,NTS)、18SrDNA、内转录间隔区1(Internal Transcribed Spacer,ITS1)、5.8SrDNA、内转录间隔区2(ITS2) 和28SrDNA组成的串联重复序列[17].
nrDNA的非编码区为高变区,受选择压小,序列差异主要表现在该区上,在近缘类群间的系统学、杂交及居群进化研究上具有一定的应用潜力[18]. ETS重复序列较少,进化较快,并且具有很高的多态性和可变性,在遗传变异、分类及系统发育研究中具有重要的应用价值. 采用ETS序列对粟的起源与传播进行研究表明,该区能很好地检测种内及种间多态性[19]. 药水苏核外转录间隔区(ETS)的分析表明,所有斯堪的纳维亚种群都有一个自发的起源,这些种群由该物种基因定义明确的亚群组成,与来自北欧大陆邻近地区的其他自发种群关系最密切[20]. Thell等[21]根据地理位置、分类学和遗传采样,基于包括ETS的6种DNA分子标记研究,重建了东亚鼠尾草的系统关系.
丹参作为我国传统的大宗药材之一,在临床上发挥着越来越重要的作用. 目前,还未见有栽培丹参居群ETS序列的遗传多样性报道. 为了解不同栽培丹参居群的ETS序列遗传多样性,为栽培丹参资源快速鉴定奠定基础,本研究对40个栽培丹参居群的ETS序列进行了PCR扩增、序列比对及系统发育树构建,探明了不同栽培丹参居群间的系统发育关系.
HTML
-
试验材料为来自国内不同地区的40种栽培丹参居群(表 1). 本实验室前期对种子形状、大小、田间栽培或盆钵培育植物的叶片、花形、花色、收获种子等农艺性状进行了鉴定,确认均为丹参材料.
-
采用十六烷基三甲基溴化铵(Cetyltrimethyl Ammonium Bromide,CTAB)法提取丹参基因组总DNA[22],经1%琼脂糖凝胶电泳确定其纯度,提取的DNA样品置于-20 ℃冰箱中保存.
-
采用上游引物F(5′-ATAGAGCGCGTGAGTGGTG-3′)和下游引物R(5′-GACAAGCATATGACTGGATCAA-3′)[23]对丹参不同栽培居群的ETS序列进行PCR扩增. PCR扩增体系为25 μL:2×SanTaq PCR Mix 11 μL,F和R (10 μmol/L) 各1 μL,DNA模板1 μL,ddH2O 11 μL. PCR扩增程序:94 ℃预变性5 min,94 ℃ 30 s,55 ℃ 30 s,72 ℃ 30 s,35个循环,72 ℃延伸10 min. PCR产物经1%琼脂糖凝胶电泳检测.
-
以回收、纯化的PCR产物为测序反应模板,通用引物为测序引物,由四川成都生工生物技术有限公司进行测序.
-
在美国国家生物技术信息中心(National Center of Biotechnology Information,NCBI)网站上将测得序列进行BLAST同源比对确认后,使用BioEdit 7.09[24]和Vector NTI Advance 11.5.3[25]软件对40个栽培丹参居群的ETS序列和GenBank中已登录的丹参ETS序列(MG824345.1)进行比对,使用分子进化遗传分析软件MEGA 7.0进行分析及系统发育树的构建[26],使用DNASP v5进行多态性位点、遗传多样性指数和中性检测分析[27].
1.1. 试验材料
1.2. 试验方法
1.2.1. 基因组总DNA提取
1.2.2. PCR扩增
1.2.3. PCR产物测序
1.2.4. 序列数据处理
-
以丹参基因组总DNA为模板,对其ETS序列进行PCR扩增,产物经1%琼脂糖凝胶电泳检测. 结果显示,ETS扩增产物均为500 bp左右的单带,条带清晰,特异性好(图 1).

-
获得的40份丹参材料的ETS序列经比对整理后,提交GenBank获得了相应的收录号(表 2). 40个栽培丹参居群的ETS序列长为421~433 bp,GC含量为56.8%~62.8%,向GC方向偏斜. 其中,居群B-GD-V-1和W-SD-V-1的长度为421 bp,居群B-SD-V-1和W-SCHY-W-2的长度为423 bp,居群W-GZ-V-1和W-GD-V-1的长度为431 bp,居群B-JS-V-1和W-JS-V-1的长度为433 bp,其余32个栽培丹参居群ETS序列长度为422 bp;GC含量最高的是居群V-GSLX-V-2,含量最低的是居群B-JS-V-1和W-JS-V-1,其余栽培丹参居群GC含量在56.8%~62.8%范围内有细微差异(表 2).
-
通过对40个栽培丹参居群ETS序列进行排序,经Vector NTI Advance 11.5.3软件比对及人工整理,结果显示:40个栽培丹参居群ETS序列的相似度为95.5%,一致性为41.0%(图 2). 存在241个SNP变异位点,变异率高达54.8%;有205个简约信息位点,变异位点丰富.
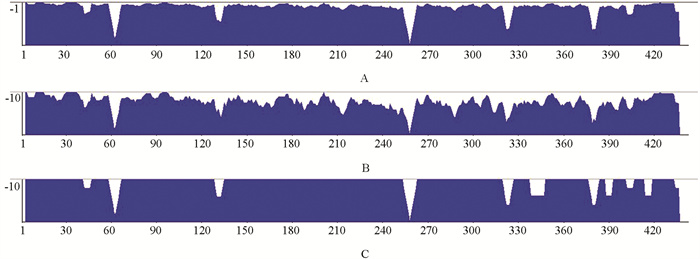
丹参ETS序列中16个居群有核苷酸变异位点,其余24个居群没有核苷酸变异. 变异位点丰富(100个以上)的居群有W-GZ-V-1,V-SD-V-1W,W-GD-V-1,B-JS-V-1,W-JS-V-1和B-SD-V-1;变异位点数中等(10~100个)的居群有W-SD-V-1,B-GD-V-1,V-JXJA-V-2和W-SCHY-W-2;变异位点较少(低于10个)的居群有V-SC-V-1,V-GD-V-1,V-JS-V-1,W-YNLJ-V-2,V-GSLX-V-2和W-HBJM-V-2(表 3).
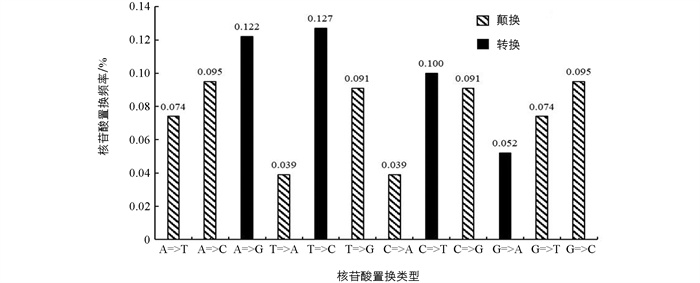
通过MEGA软件找到ETS序列的24个核苷酸替代模型,分别为HKY+I,HKY+G,T92+I,HKY,HKY+G+I,TN93+I,T92+G,TN93,T92,TN93+G,T92+G+I,TN93+G+I,JC+G,JC+I,K2+G,K2+I,JC,JC+G+I,K2,K2+G+I,GTR+I,GTR+G,GTR和GTR+G+I. 根据贝叶斯准则(Bayesian Information Criterion,BIC),其中HKY+I模型的BIC分数最低,为ETS序列的最佳替代模型. 栽培丹参居群ETS序列转换率和颠换率分别为40.1%和59.9%,其转换率低于颠换率(图 3).
-
通过DNASP软件,对40个栽培丹参居群的遗传多样性进行分析获得16种单倍型,单倍型多样性指数(Haplotype Diversity,Hd)为0.640,核苷酸多样性指数(Nucleotide Diversity Index,π)为0.112,单倍型多样性方差(Variance of Haplotype Diversity,Vh)为0.008,单倍型多样性标准差(Standard Deviation of Haplotype Diversity,Sh)为0.089. 中性检验表明ETS序列在p>10%水平上均不具有统计学意义,符合中性进化模型.
-
利用最大似然法对ETS序列的遗传距离进行估算. 结果表明:在标准误估计值为0.052的情况下,40个栽培丹参居群的总平均遗传距离为0.151. 24个没有核苷酸变异位点的居群用C24表示. 16个具有SNP指纹的栽培丹参居群及其之间的遗传距离表明:居群W-SD-V-1与居群B-JS-V-1和W-JS-V-1的遗传距离最大,达到0.648,说明居群W-SD-V-1与居群B-JS-V-1和W-JS-V-1的亲缘关系较远,居群间遗传多样性高;而居群W-JS-V-1与B-JS-V-1的遗传距离最小,数值为0,说明两个居群间的亲缘关系较近,居群间遗传多样性低(表 4).
-
根据获得的栽培丹参居群ETS序列,以GenBank下载的湖北鼠尾草ETS序列(MG824345.1)为外群,用邻接法(NJ)、最大似然法(ML)和最大简约法(MP)3种方法构建系统进化树,考察其系统发育关系. 结果表明:3种进化树的拓扑结构大致相同. 最大似然法和邻近连接法构建的进化树显示,40个栽培丹参居群聚类在2个系支上,相同的24个居群聚类在一个系支上,另外相同的、具有变异位点的16个居群(表 3)聚类在另一系支上(图 4A,B);而最大简约法构建的进化树显示,40个栽培丹参居群聚在2个系支上,27个居群聚类在一个系支上,另外13个居群聚类在另一系支上(图 4C). 可见,最大似然法和邻近连接法更适合丹参ETS聚类分析.

2.1. 不同栽培丹参居群ETS序列的PCR扩增
2.2. 丹参ETS序列特征
2.3. 不同栽培丹参居群ETS序列的核苷酸变异分析
2.4. 遗传多样性分析
2.5. 遗传距离分析
2.6. 基于丹参ETS序列的系统发育关系
-
ETS序列进化较快且具有较高的多态性和可变性,在遗传变异、分类及系统发育研究中有着重要的用途. 由于ETS序列难以找到合适的通用引物扩增整个区域,所以在系统学研究中的例子比较少[28]. 本研究采用应用于辣椒的ETS引物[23]首次成功扩增了丹参的ETS序列,其长度为421~433 bp,GC含量为56.8%~62.8%;发现40个栽培丹参居群的ETS序列共有241个SNP变异位点,变异率高达54.8%,有205个简约信息位点,与同属唇形科植物的ETS研究结果相似[29-30];还发现基于ETS序列的SNP指纹可鉴定14个栽培丹参居群,但不足以区别所有栽培丹参居群,因此笔者认为准确可靠的分子鉴定可能依赖于从多种DNA分子标记上寻找可靠的复合分子标记.
本研究通过MEGA软件找到ETS序列的最佳核苷酸替代模型为HKY+I,发现核苷酸转换率(40.1%)低于颠换率(59.9%),而相对较高的核苷酸置换率说明丹参种间和种内具有较高的遗传变异性[31];丹参ETS序列的单倍体多样性指数Hd为0.64(>0.5),表明栽培丹参的遗传资源丰富,这与基于EST-SSR标记的丹参遗传多样性研究结果一致[16]. 中性检验结果表明ETS序列在p>10%的水平不具有统计学意义,符合中性进化模型,表明栽培丹参居群近期并没有发生扩张现象.
基于ETS序列的最大似然法、邻近连接法和最大简约法聚类结果表明,40个栽培丹参居群聚类在两个系支上,3种进化树的拓扑结构大致相同,最大似然法和邻近连接法聚类结果一致,更适合丹参ETS聚类分析. 研究结果为进一步的栽培丹参分子系统学研究奠定了基础.
 DownLoad:
DownLoad: