




-
开放科学(资源服务)标志码(OSID):

-
叶绿体是绿色植物进行光合作用最重要的细胞器,是许多基本物质和次生代谢物合成的重要场地[1]. 叶绿体蛋白虽然绝大多数是由核基因编码,但也有100多种蛋白是由叶绿体基因组(cpDNA)编码的. cpDNA序列长度为130~150 kb,具有典型的双链环状结构,由1个大单拷贝区(LSC),1个小单拷贝区(SSC)和两个反向重复区(IR)组成. 大多数的cpDNA是母系遗传,但也有一部分植物是父系遗传或双亲遗传. 相对于核基因组的复杂多样性,cpDNA结构简单,序列高度保守,不同物种或同一物种不同个体间仅存在着局部区域的序列变异,因此cpDNA更易解析,更有利于研究植物的分类与进化[2-4].
甘薯是我国重要的粮食作物、饲料作物和食品加工业、化工业的原料作物[5]. 叶菜型甘薯是一类以鲜幼嫩茎叶作蔬菜用的甘薯品种,与普通甘薯相比,其茎叶产量较高[6]. 叶绿体基因组的完整解析是准确研究植物系统进化关系、发掘基因功能的更有效而可靠的手段[7]. 叶菜型甘薯的父本或母本往往来源于普通甘薯,其叶绿体基因组序列与普通甘薯有什么差别目前尚未清楚,甘薯种间叶绿体基因组之间存在多少碱基差异目前也不清楚. 本研究以叶菜型甘薯‘福菜薯18号’为材料,通过序列拼接获得完整的叶绿体基因组,并利用生物信息学方法进行分析,为叶菜型甘薯的亲缘关系和甘薯种间关系奠定基础.
HTML
-
以叶菜型甘薯‘福菜薯18号’为试验材料,其嫩梢幼叶于2021年7月采自‘福菜薯18号’甘薯组培苗.
-
利用DNA提取试剂盒(南京诺维赞生物科技有限公司)提取甘薯组织总DNA[8],用1.5%琼脂糖凝胶电泳和Nanodrop 2000检测甘薯总DNA的质量和完整性,DNA质量合格后进行上机测试.
二代测序实验流程按照BGISEQ-500的标准程序执行,样品基因组DNA检测合格后,用超声波法将DNA片段化,然后对片段化的DNA进行纯化,末端修复,3′端加A,连接测序接头,构建测序文库,文库质检合格后用BGISEQ-500平台进行测序.
三代测序采用Oxford Nanopore Technologies公司的建库试剂盒进行测序文库构建,文库检验合格后上机测序.
-
使用Fastp软件对原始数据进行过滤,去除其中的接头序列及低质量序列,获取高质量的序列数据,按参考物种的叶绿体基因组序列进行组装,得到叶绿体基因组序列组装结果[9]. 使用minimap2将三代测序reads比对NCBI旋花科所有叶绿体基因组数据,提取比对长度大于5 000 bp的reads用于后续组装[10]. 使用bowtie2将二代测序reads比对广州佰数生物科技有限公司自建的叶绿体基因组数据库,将比对上的reads用于后续组装[11]. 组装软件使用Unicycler version:v 0.4.8,将上述提取到的叶绿体候选三代和二代reads用于叶绿体基因组组装[12].
使用GeSeq软件对叶绿体基因序列进行注释[13];利用tRNAscan-SE在线网站对tRNA进行注释[14],利用RNAmmer 1.2 Server(
http://www.cbs.dtu.dk/services/RNAmmer/ )对rRNA进行注释,经人工修正后获得最终的注释结果;最后使用OGDRAW(https://chlorobox.mpimp-golm.mpg.de/OGDraw.html )软件生成基因组物理图谱[15]. -
根据Sharp等[16]计算方法对组装好的叶菜型甘薯叶绿体基因组序列进行密码子偏好性(Relative Synonymous Codon Usage,RSCU)值统计和分析. 采用MISA软件(
http://pgrc.ipk-gatersleben.de/misa/ )对组装好的叶菜型甘薯叶绿体基因组序列进行微卫星扫描[17],利用Tandem repeats finder v 4.04软件检测串联重复序列,默认参数参考文献[18].由于叶绿体基因组组装过程中发现有两种类型的叶绿体,即叶绿体基因组在SSR区域具有正反两种方向的结构,因此利用dottup软件对两种类型的叶绿体基因组进行共线性分析[19].
-
从NCBI数据库下载番薯属Ipomoea trifida(NC_034670),Ipomoea cordatoteiloba(NC_041204),Ipomoea lacunosa(NC_037912),Ipomoea cynanchifolia(NC_041203)和Ipomoea ramosissima(NC_041205) 等45个种植物叶绿体基因组序列与叶菜型甘薯进行聚类分析,利用其与两个外群物种全叶绿体序列构建进化树. 使用软件mafft(默认参数)进行序列比对[20],而后利用fasttree软件构建ML进化树[21].
-
从NCBI数据库下载8个甘薯叶绿体基因组数据,将9个基因组序列用mafft对比后,以‘福菜薯18号’叶绿体基因组为参考序列进行SNP和Indel分析.
1.1. 试验材料
1.2. DNA的提取及测序
1.3. 基因组组装与注释
1.4. 叶绿体基因组数据分析
1.5. 番薯属叶绿体基因组比较
1.6. 甘薯种间叶绿体基因组SNP和Indel分析
-
叶菜型甘薯的cpDNA序列全长为161 387 bp,由大单拷贝区域(LSC,87 597 bp),小单拷贝区域(SSC,12 052 bp)及两个反向重复区域(IRA和IRB,30 869 bp)4个部分构成(图 1). 组装注释好的叶绿体基因序列提交至GenBank,获得序列登录号OM808940. 基因注释结果表明:叶菜型甘薯cpDNA具有132个功能基因,包括87个蛋白编码基因、8个rRNA基因和37个tRNA基因. 其中,18个基因在IR区域复制,包括7个蛋白编码基因(ycf1,ycf2,ycf15,ndhB,ndhH,rps7和rps15),7个tRNA基因(trnI-CAU,trnL-CAA,trnV-GAC,trnI-GAU,trnA-UGC,trnR-ACG和trnN-GUU)和4个rRNA基因(rrn4.5,rrn5,rrn16和rrn23). 叶菜型甘薯cpDNA总的GC质量分数为37.54%,AT质量分数为62.46%.
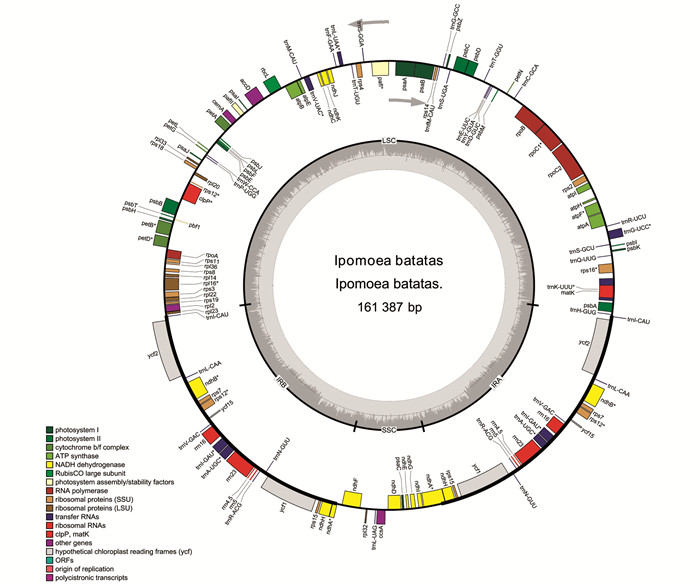
经过统计分析,叶菜型甘薯cpDNA中20个基因包含内含子,其中,11个蛋白编码基因和7个tRNA基因含有1个内含子,2个蛋白编码基因(pafI和clpP)含有2个内含子(表 1). rps12有2个拷贝,每个拷贝具有3个外显子,且两个拷贝共享第1个外显子,第1个外显子位于LSC区域,另外2个外显子位于IR区域.
-
对于不同的生物体蛋白质结构组成,即使编码氨基酸的密码子相同,但是对于氨基酸的同义密码子使用频率却是不相等的,这种同义密码子使用频率的不相等就是密码子偏好性(RSCU). 叶菜型甘薯cpDNA中RSCU值大于1.00的密码子为32个,其中大多数以A或T结尾,仅3个以G结尾(ATG,TTG,TGG). 编码亮氨酸(Leu)的密码子数量最多,为2 950个,占比10.35%;半胱氨酸(Cys)出现的次数最少,为332,占比1.16%(表 2). 这与大多数被子植物叶绿体基因组密码子使用偏好一致.
-
SSR(Simple Sequence Repeats) 是一类由1~6个核苷酸为重复单位组成的长达几十个核苷酸的串联重复序列,每个SSR两侧的序列一般是相对保守的单拷贝序列. 从叶菜型甘薯叶绿体基因组中共鉴定到54个SSR位点,其中,单核苷酸、二核苷酸、三核苷酸、四核苷酸、五核苷酸和六核苷酸重复分别有32,4,3,11,2和2个(表 3),且32个单核苷酸重复均由A或T组成.
-
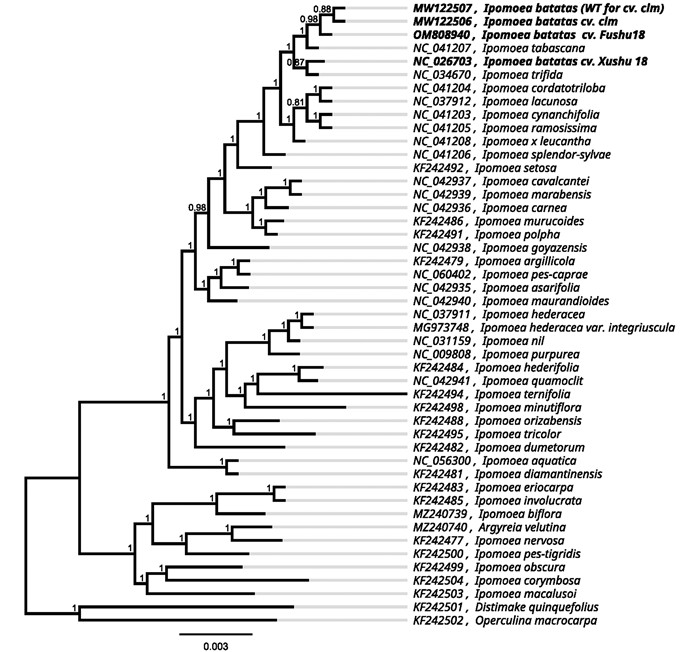
从NCBI上下载旋花科甘薯近缘种植物的叶绿体基因组序列,对全基因组序列的共有蛋白编码基因进行聚类分析,并以Distimake quinquefolius和Operculina macrocarpa为外类群. 结果显示,叶菜型甘薯与普通甘薯‘clm’和甘薯四倍体野生种Ipomoea tabascana聚为一类,普通甘薯‘徐薯18号’和甘薯二倍体野生种Ipomoea trifida聚为一类,同时聚为一个大类. 旋花科的黄毛银背藤也与番薯属植物聚为一类,两个外群植物Distimake quinquefolius和Operculina macrocarpa聚为一类,与番薯属植物分为两个进化支(图 2).
-
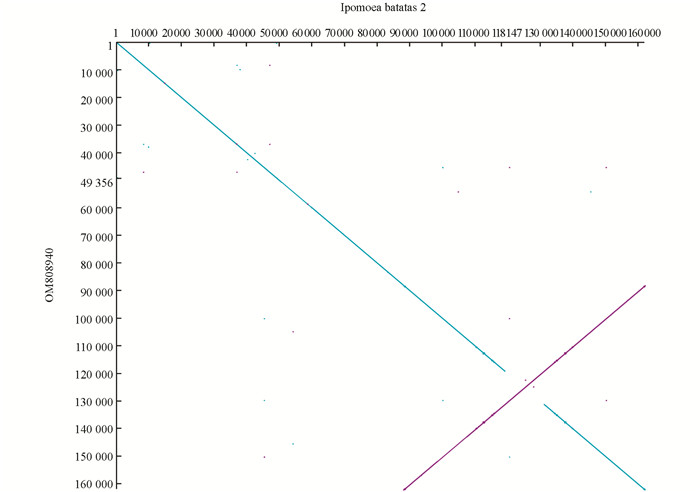
由于叶绿体基因组组装过程中发现有两种类型的叶绿体,即叶绿体基因组在SSR区域具有正反两种方向的结构,利用dottup软件对两种类型的叶绿体基因组进行共线性分析,其中蓝色是正向共线性,紫红色是反向共线性(图 3).
-
以‘福菜薯18号’为对照,对甘薯品种间叶绿体基因组进行种内SNP分析,发现有199个SNP位点,其中有118个位点位于编码区中,编码区包含matK,rpoC2,psaB,accD,psbL,rps8,ycf1,ycf2,ndhB,ndhC,ndhE,ndhF,ndhH等基因,ycf1和ycf2具有较多的SNP位点,其余位于非编码区中. ‘福菜薯18号’叶绿体基因组与其他甘薯相比,有121个片段缺失,其中有37个属于SSR位点缺失;有146个片段插入,其中有66个属于SSR位点插入;另外还有7个长片段替换(数据略).
2.1. 叶菜型甘薯叶绿体基因组的基本特征
2.2. 叶菜型甘薯密码子的偏好性
2.3. 叶菜型甘薯cpDNA简单重复序列组成
2.4. 系统进化分析
2.5. 叶菜型甘薯两种类型的叶绿体基因组
2.6. 甘薯品种间叶绿体基因组的SNP和Indel分析
-
20世纪70年代末,双脱氧终止法标志着第一代测序技术的诞生,实现了对DNA序列的测序与分析,由于这种方法测序通量低、自动化水平差等缺点,限制了其在转录组学和基因组学的发展. 第二代测序是边合成边测序,通过捕捉末端新合成的碱基来获得待测DNA片段的序列,实现了高通量和自动化测定,极大地提高了测序速度,但由于二代测序包含PCR扩增等过程,可能会引入模板迁移等假阳性,而且二代测序读长普遍较短,也限制了其应用. 三代测序技术以PacBio公司的单分子实时测序技术(Single Molecule Real Time Sequencing,SMRT-seq)和Oxford Nanopore Technologies的纳米孔单分子测序技术为代表,与前两代测序技术相比,其最大的特点就是单分子实时测序,测序过程无需进行PCR扩增,可以实现长片段序列测定,但缺点是通量相对较小,测序成本较高[22-23]. 本文为提高叶菜型甘薯叶绿体基因组测定序列的准确性,采用了二代和三代测序数据相结合,克服了测序技术本身的不足,保证序列拼接组装的准确性.
甘薯属于旋花科番薯属植物,本研究从NCBI上下载了45个番薯属植物的叶绿体基因组数据,包括普通甘薯‘徐薯18号’等叶绿体基因组数据并进行序列对比,并以Distimake quinquefolius和Operculina macrocarpa为外类群构建系统进化树,结果显示,叶菜型甘薯与甘薯四倍体野生种Ipomoea tabascana和甘薯二倍体野生种Ipomoea trifida聚为一类. Srisuwan等[24]通过细胞遗传学方法认为甘薯栽培种与野生种Ipomoea trifida具有更为密切关系,Ipomoea trifida是甘薯栽培种和四倍体Ipomoea tabascana的祖先,与叶绿体基因组进化相一致. 有研究表明,番薯属植物Ipomoea purpurea与其近缘种Ipomoea alba和Ipomoea nil在花青素合成基因中有约1%的核苷酸多态性,符合分子进化的标准中性模型[25]. 甘薯种内SNP分析结果表明,甘薯叶绿体基因组总长度有所差异,‘徐薯18号’的叶绿体基因组长度为161 303 bp[26],甘薯‘clm’及其对照分别为161 393 bp和161 429 bp[27],甘薯叶绿体基因中存在单核苷酸突变,也存在长片段替换、缺失和插入,这些碱基差异有些是位于编码区基因matK,rpoC2,psaB,accD,rbcL中,其中matK和rbcL是植物DNA条形码的核心序列[28],另外一些编码基因是否可以成为番薯属的DNA条形码有待更多数据的支持.
 DownLoad:
DownLoad: