


 下载:
下载:
-
土壤颗粒随水迁移常引发土壤退化、农田面源污染等一系列生态与环境问题[1].其中土壤团聚体的破碎被认为是土壤颗粒随水迁移发生的第一步,也是关键一步[2],因此,控制土壤团聚体破碎程度是控制土壤颗粒水迁移强度直接而有效的手段[3].已有研究表明,土壤颗粒间的相互作用是团聚体破碎的内在驱动力[3].土壤颗粒间的水合斥力[4]、静电斥力和分子引力产生的颗粒间的压强可达到102~103atm[5],而雨滴打击力等外部作用力只能产生1~3 atm的作用压强[6].因此,相比于经典侵蚀理论中的各种外力作用,土粒间的内部作用力才应是土壤团聚体破碎的决定因素[3].
根据胶体颗粒相互作用的DLVO理论,颗粒间的静电斥力受颗粒周围电场的控制[7-8],而离子界面反应特征决定着土壤土粒周围的电场强度[3].经典DLVO理论只考虑了离子界面反应中离子电荷价的贡献,该理论中同价离子对土壤颗粒相互作用力的影响相同,但大量实验表明同价态不同离子在土壤颗粒相互作用中表现出了强烈的离子特异性效应[9].例如,K+体系团聚体稳定性强于同浓度下的Na+体系,而后者的分散性更强;含交换性Na+较多的盐碱土结构差,肥力低,而富含K+的紫色土肥力较高.
现有理论认为离子特异性效应的产生机制主要来自于离子大小、离子水合和色散力等作用[10].但这些因素都只有在较高浓度(大于0.1 mol/L)电解质条件下才能发生作用,并且随着电解质浓度的降低而逐渐减弱[11].然而,有研究发现低浓度电解质条件下仍然存在着非常强的离子特异性效应,且该效应随电解质浓度的降低变得更加明显[12-13]. Liu等人[14]发现,在土壤胶体颗粒强外电场作用下(108~109 V/m),离子的量子涨落被急剧放大而发生强烈的非经典极化作用(其极化率可达到经典极化率的成百甚至上千倍).离子间非经典极化的差异导致这些离子与土壤颗粒界面反应间的差异,从而导致土壤颗粒间相互作用的差异. Li等人[3]研究发现,离子的非经典极化会对土壤流失强度产生影响. Du等人[15]定量计算了一些一价金属阳离子的非经电极化率,发现这些一价金属阳离子的非经典化率差异巨大.比如在10-4 mol/L电解质浓度下,Cs+,K+和Na+的非经电极化率分别为2209,1702和417.9Å3.非经电极化率的巨大差异本质上反映了这些同价离子核外电子的能量和量子状态的巨大差异,而核外电子的能量和量子状态的巨大差异将使这些同价金属离子在土粒表面表现出不同的界面反应特征.
本研究通过K+和Na+两种离子在蒙脱石上的界面反应特征来表征金属离子界面反应如何影响粘土的水迁移.本研究的主题不是土壤中的离子构成如何影响土壤物质迁移,而是离子界面反应如何影响了土壤物质的水迁移.基于此,这里仅以K+和Na+两种离子作为本研究的指示离子.由于土壤组成十分复杂,使得包括静电作用力在内的各种作用力的定量计算都非常困难,因此,本研究采用蒙脱石作为供试材料.
全文HTML
-
本研究所用蒙脱石购买自内蒙古物华天宝矿物资源有限公司.采用Li等[16]人所建立的物质表面电化学性质参数联合测定方法测得蒙脱石矿物阳离子交换量和比表面积分别为84.8 cmol/kg和716 m2/g.
为定量表征不同浓度与类型电解质条件下,粘土颗粒相互作用中的离子特异性效应,本研究将蒙脱石分别制成Na+饱和样和K+饱和样.制备方法如下:称取1 kg蒙脱石,加入0.5 mol/L的NaNO3溶液5 L,搅拌分散24 h后,离心弃去上清液(4 000 r/min,5 min),重复操作3次;之后,加入去离子水5 L搅拌24 h,离心弃去上清液以洗去多余盐分,重复2~3次. 70 ℃下烘干,获得Na+饱和样,过筛取0.5~2 mm团聚体用于模拟降雨实验. K+饱和样的制取采用KNO3溶液,方法同上.
-
将粒径为0.5~2 mm的Na+/K+饱和团聚体平铺于5 cm × 5 cm的土槽内,厚度为3 cm,坡度为30°.土槽末端安放孔径2 mm的阻拦网,土槽下端导流槽可汇总并收集随水流出的粘土颗粒.将土槽安放在针滴式滴水器下,针尖出水口距离土槽中样品表面3 cm,以尽量降低水滴对样品的打击作用,水流通量为25 mL/(cm2·min).水中分别含NaNO3或KNO3电解质,浓度分别为0.000 1,0.001,0.01,0.1和1 mol/L,水源温度为25 ℃.从水流产生开始计时,初期每2 min收集一次流出液,10 min后,每5 min收集一次,实验时间为90 min.将收集的悬液在105 ℃条件下烘干并称重.获取粘粒各时间段的流失量和累积流失量.实验重复3次.
-
近年的相关研究已经指出,相同价数的不同阳离子由于核外电子构象的差异,使得这些离子表现出完全不同的界面反应特征,而不同的反应特征集中体现在这些离子具有不同的非经典极化效应[16].同价离子非经典极化的差异必定引起这些离子屏蔽粘粒周围电场能力的差异,进而引起粘粒表面电位和颗粒间作用力的差异.所以,计算粘粒表面电位和粘粒间的静电作用力时,需考虑离子不同的表面反应特征.
对于1:1型电解质体系,考虑离子非经典极化效应的颗粒表面电位可由下式计算[14]:
其中:
式中:φ0为蒙脱石颗粒表面电位(V);R为气体常数(J/(mol·K));T为绝对温度(K);F为法拉第常数(C/mol);Z为阳离子价数;γ为因阳离子非经典极化而产生的有效电荷数[13];CT为蒙脱石阳离子交换量(mol/g);S为蒙脱石颗粒比表面积(dm2/g);c0为本体电解质浓度(mol/L);κ为德拜参数(dm-1),其倒数1/κ表示胶体颗粒扩散双电层厚度;ε为水的介电常数,取值为8.9×10-10 C2/(J·dm).
对于1:1型电解质体系,考虑离子非经典极化效应的颗粒间静电斥力的大小可以通过下式进行计算[16]:
式中:PEDL为相邻两颗粒间的静电排斥压强(atm);d为两颗粒间的距离(dm);φ(d/2)为相邻两个蒙脱石颗粒双电层重叠中点处的电位值(V),其可以通过下式计算[13]:
1.1. 材料及蒙脱石团聚体制备
1.2. 实验方法
1.3. 考虑离子不同界面反应特征的粘粒表面电位和粘粒间静电排斥力计算
-
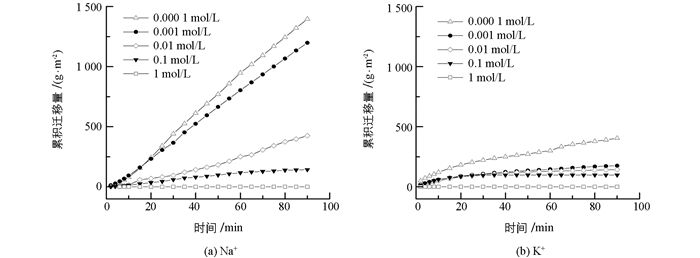
本研究首先测定了不同Na+,K+浓度下蒙脱石颗粒的累积迁移量随时间的变化关系.从图 1可知:1)电解质浓度高达1 mol/L时,Na+,K+体系下粘粒累积迁移量均为0 g/m2;当电解质浓度低于0.1 mol/L时,随着电解质浓度的降低,粘粒累积迁移量逐渐增大. 2)在整个实验过程中,Na+存在时的粘粒累积迁移量均显著大于K+存在时的迁移量.例如,当电解质浓度为0.000 1 mol/L时,Na+和K+体系在90 min降雨时粘粒累积迁移量分别为1443.0和404.4 g/m2,前者是后者的3.6倍.实验结果表明,粘粒的水流迁移量存在明显的离子特异性效应. 3) Na+,K+两个体系中的粘粒累积迁移量随时间的变化趋势有所不同,其拟合函数关系列于表 1.由表 1可知,Na+存在时,粘粒累积迁移量随时间呈线性增长,粘粒的水流迁移速率是一个常数,且随着电解质浓度的降低,粘粒迁移速率逐渐增大.这表明在实验的给定时间范围内,粘粒的水流迁移量是稳定且持续的. K+体系中,粘粒累积迁移量与时间为非线性关系,随时间的增加,粘粒累积迁移量将趋于一个极限最大值,且随电解质浓度增加,粘粒水流迁移量的极限最大值迅速降低.这也表明,当K+存在时,初期阶段粘粒水流迁移的强度最大,随时间的增加水流迁移的强度逐渐降低,并在水流迁移发生一定时间后粘粒迁移不再发生.
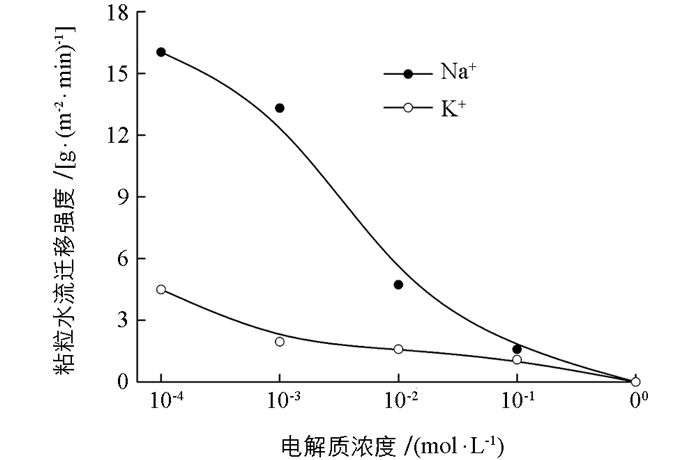
如果将平均累计水迁移速率看成是水流迁移强度,两种离子分别存在时颗粒的水流迁移强度与电解质浓度的关系如图 2所示.由图 2可知,Na+与K+体系间的差异随着离子浓度降低而增大,在高浓度时,如1 mol/L时,两者均未发生水流迁移.当浓度降低到0.001 mol/L时,Na+与K+体系的粘粒水流迁移强度分别为13.3和2.0 g/(m2· min),前者是后者的6.7倍. K+体系水流迁移强度随离子浓度增加非常缓慢,而Na+体系增加迅速.
-
金属离子在粘粒表面不同的界面反应特征必定影响粘粒周围的电场强度,进而影响粘土团聚体中粘粒之间的静电排斥压强,从而影响粘土团聚体的稳定性和孔隙状况,最终影响粘粒随水迁移的强度.而经典的DLVO理论无法解释两种同价离子之间颗粒间相互作用力的差异. Na+与K+的离子大小、离子水合和色散力效应都有一定差异[10],这些差异也可能带来粘粒间相互作用力的不同.但是,不论是离子体积、离子水合体积还是色散力作用,这些差异均只能在较高电解质浓度环境中才能表现出来,即只有在高浓度下才能表现出两种离子下的粘粒相互作用力和粘粒水迁移强度的差异.然而,图 2表明,水迁移强度的差异在低电解质浓度下最大,高电解质浓度下反而最小.所以,离子大小、离子水合和色散力效应不是两种离子存在时水粘粒水迁移强度差异的原因.
蒙脱石颗粒表面电场强度可达1.6×108 V/m,在如此强的电场中,粘土颗粒表面的金属离子发生了非经典极化[15].当处于相同电场时,K+核外电子云比Na+更易发生形变,因此K+非经典极化作用更强,其屏蔽粘粒周围电场的能力也更强,从而降低了蒙脱石团聚体中粘粒之间的静电排斥力,增强了团聚体的稳定性,降低了粘粒的水迁移强度.那么在这两种离子体系中,蒙脱石团聚体中颗粒间的作用力差异到底有多大呢?
-
表(2)是考虑离子界面反应中非经典极化效应而得到的两种离子在不同电解质浓度下蒙脱石颗粒的表面电位.从表(2)可以看出,Na+,K+不同的界面反应特征(两种离子在界面上不同的极化强度)使得同一蒙脱石材料表现出不同的表面电位值,而且K+存在时的表面电位远远低于Na+存在时的表面电位.这也预示,K+存在时颗粒间的静电斥力远远低于Na+存在时颗粒间的静电斥力.
-
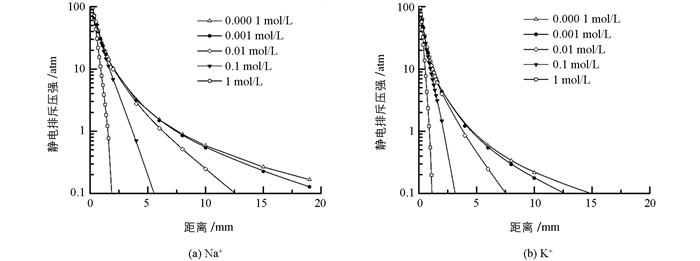
图 3是考虑离子界面反应中非经典极化效应而得到的两种离子在不同电解质浓度下蒙脱石颗粒间的静电排斥力分布. 图 3的确表明,K+存在时颗粒间的静电斥力远远低于Na+存在时颗粒间的静电斥力.
-
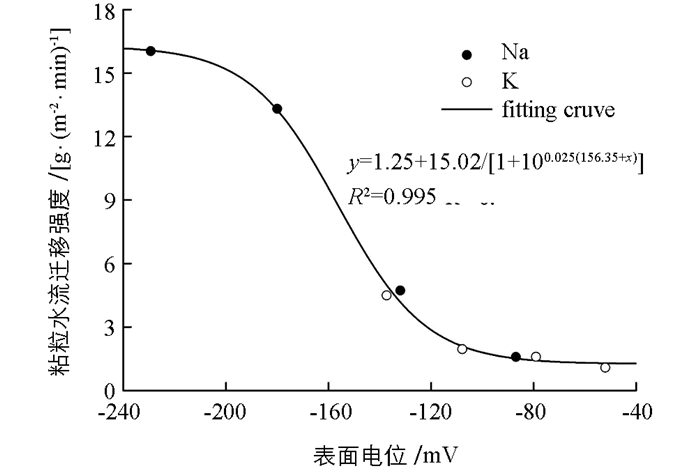
根据前面分析,两种阳离子界面反应特征的差异带来了体系静电效应的差异(可用表面电位和静电排斥力表征),这种差异最终带来粘粒水迁移强度的差异.因此,如果静电效应才是引发粘土水流迁移的内在原因,那么用表面电位代替图 2中横坐标的电解浓度重新进行作图,图 2中两种离子对应的两条曲线将可以统一地用同一条曲线表达,如图 4所示. 图 2中粘粒水流迁移强度与电解质浓度之间的关系,K+,Na+两种离子遵循完全不同的关系,但在图 4中,两种离子的确遵循同一个幂函数关系.这表明:1)引起粘粒水迁移发生强度差异的本质原因不是电解质浓度的不同,而是粘粒周围的电场强度的不同(用表面电位间接反映);2)基于离子非经典极化作用的金属离子界面反应,通过其对粘粒周围电场强度的影响来影响粘粒的水迁移强度.可以预期,在不同离子体系中,该效应对粘土颗粒水迁移的影响方面具有普遍性.
根据实验数据得到的粘粒水迁移强度与表面电位之间的幂函数关系为:
式中:x为蒙脱石颗粒表面电位;y为粘粒水迁移强度.
粘土体系的电场可以从多个方面影响粘粒之间的相互作用,其中一个最基本的影响就是电场通过影响粘粒间的静电排斥力来影响粘粒间的相互作用.因此基于上述讨论,我们自然地会问:粘粒体系的电场是否通过影响颗粒间的静电排斥力来影响粘粒的水迁移呢?
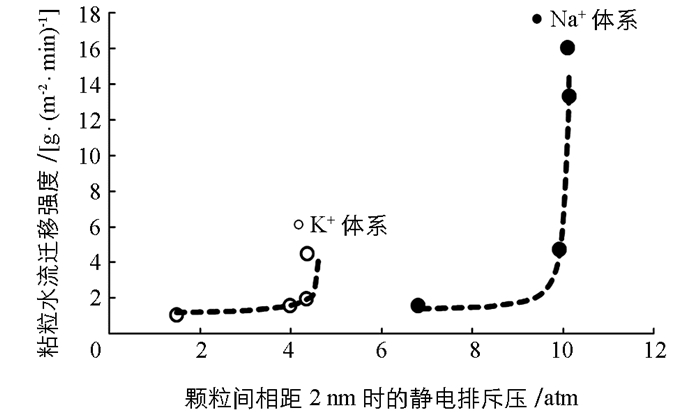
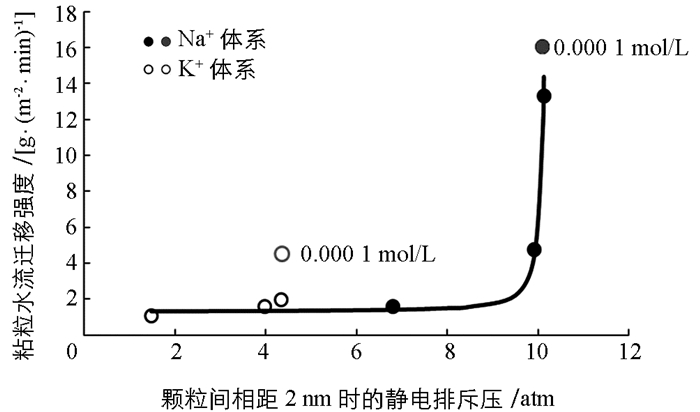
图 3已经给出了两种离子体系中不同电解质浓度下蒙脱石颗粒间的静电排斥力与颗粒间距离变化间的关系,在这里我们以颗粒间相距2nm时的静电排斥压作为横坐标,以粘粒水迁移强度为纵坐标作图,探讨粘粒水迁移强度与静电排斥力之间的关系.作图结果如图 5所示.
从图 5可以看出,不论是K+体系还是Na+体系,低的静电排斥压始终对应低强度的粘粒迁移,因此颗粒间的静电排斥压必定是粘粒水迁移的重要推动力.但与图 3不同的是,图 4似乎表明两种离子体系的“迁移强度-排斥压关系”似乎更适合于用两个不同的函数关系来表达.这表明,静电场除了通过颗粒间静电排斥压来作用与粘粒水迁移外,该静电场还可能产生其它效应来影响粘粒水迁移.
如果我们用一个统一的函数来表达迁移强度与静电排斥压之间的关系,发现如下函数能够很好地表达除0.000 1 mol/L这一低浓度电解质之外的迁移强度与静电排斥压之间的关系:
式中:p为颗粒间相距2 nm时的静电排斥压.
图 6就是根据方程(7)绘制的“迁移强度(y)-排斥压关系(p)”之间的关系曲线(实线)与实验数据的比较.该图表明,两种离子都是在实验的最低电解质浓度(0.000 1 mol/L)时出现严重偏离有方程(7)给出的理论预测.比如,对于K+体系而言,当电解质浓度为0.000 1 mol/L时粘粒迁移的理论计算值为1.344 g/(m2·min),而实验值为4.493 g/(m2·min);而该电解质浓度下Na+体系的理论计算与实验值分别为11.11和16.03 g/(m2·min).而其他电解质浓度下两种离子的相关理论计算值与实验值则相当吻合.表明理论计算值都大大低于实验测定值.这一结果预示着我们低估了颗粒间的排斥压.换一句话说,在低电解质浓度下,体系的静电场除了产生颗粒间的静电排斥压外,还可能产生其它的未知的排斥压.另一方面,由于电解质浓度最低对应的是各体系最强的电场(从表 2的表面电位值可以间接地反应),所以这个未知的排斥压应该与静电场强度成正相关关系.显然因静电场产生的粘粒间的水合排斥压可能是这个未知的排斥压.原因是,静电场越强,水偶极子与颗粒表面间的引力就强,颗粒表面吸附的水分子层越厚,颗粒间的水合排斥就越强.但由于这涉及比较复杂的水合作用机制问题,所以针对这一推论,我们将在以后工作中做深入研究.
2.1. 不同Na+,K+浓度下蒙脱石颗粒的水流迁移强度
2.2. 基于Na+,K+不同界面反应特征的粘粒相互作用力研究
2.2.1. 两种离子体系中蒙脱石颗粒的表面电位
2.2.2. 两种离子体系中蒙脱石颗粒的静电排斥力分布
2.3. 界面反应对蒙脱石水流迁移强度的影响
-
离子界面反应强烈影响着粘土颗粒随水迁移的离子特异性效应.颗粒表面电场是控制团聚体稳定性的决定因素,而表面电场则通过离子界面反应所控制.电场强度越强,颗粒间的静电排斥压越高,粘粒水迁移强度就越大.例如实验中Na+离子-表面相互作用弱于K+,因此Na+体系中蒙脱石颗粒表面电场强于k+体系,相应地Na+体系中粘粒间静电排斥力远强于K+体系,从而导致Na+体系中粘土颗粒迁移强度远远大于同浓度下的K+体系,而且两体系间粘粒水迁移强度的差异随着电解质浓度的降低而增大.
研究发现离子不同的界面反应特征决定了相同电解质浓度下粘土团聚体中的电场强度,而且正是因为团聚体中的不同电场强度决定了粘粒之间排斥压的强度的差异,最终带来了粘粒随水迁移强度的差异.只有当离子界面反应中考虑这种差异时,理论预测结果才能与实验测定值吻合.而经典颗粒相互作用理论由于没有考虑离子界面反应中的离子特异性效应而不能正确预测颗粒间的相互作用力强度,从而使粘粒水流迁移强度的实验结果与理论预测相差悬殊.另一方面,在0.000 1 mol/L的低浓度条件下,颗粒表面电场更强,水偶极子在颗粒表面出现强吸引力,颗粒表面吸附的水分子层越厚,颗粒间的水合排斥就越强.因此,在低浓度条件下,颗粒水合排斥力对颗粒间相互作用有着重要影响而不可忽略.