


 下载:
下载:
-
静高压作为一种极端条件,为材料研究提供了一种重要的途径[1].氮化金属材料的研究比较重要,著名的GaN半导体材料是蓝光LED的主要发光材料[2].高压下第二主族元素氮化物M3N2(M=Be,Mg,Ca,Sr,Ba)的性质研究是一个重要的研究领域. 2009年RÖMER等人[3-4]理论预测了几种未知的高压相Ca3N2和Mg3N2.随后吉林大学郝健等人[5-6]从实验和理论方面研究了两种氮化物的高压性质,理论研究结果与RÖMER的结果是一致的.文献[5-6]中γ相的Ca3N2没有被XRD实验观测到,但是,2011年德国科学家BRAUN等人[7]在实验中合成了新的高压相γ-Ca3N2.近年来,人们对Ca3N2的研究主要集中在用其作为发光薄膜材料上[8],而并未对γ-Ca3N2晶体有深入的研究.本文基于第一性原理方法,研究了新的高压相γ-Ca3N2.主要计算了材料在常压下的晶体结构,预测了高压下的电子结构和光学性质,为进一步的研究提供参考.
全文HTML
-
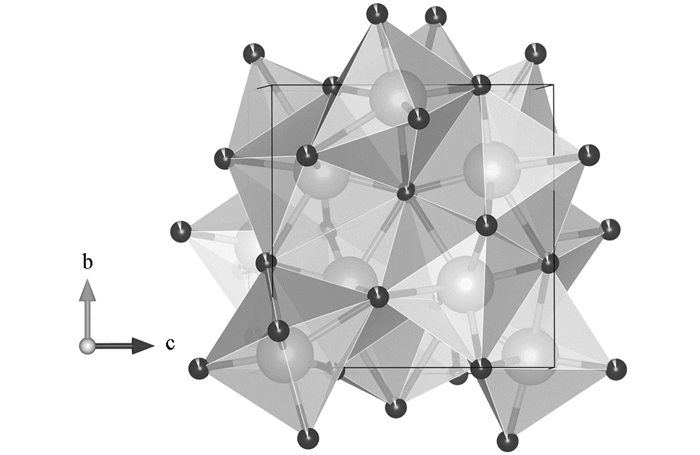
对高压相γ-Ca3N2晶体在0~115 GPa压力区间的性质进行了计算.计算过程均采用周期性边界条件,电子间的交换关联函数采用广义梯度近似(GGA)的PBE [9](Perdew-Burke-Ernzerhof)方法; 晶体中电子的波函数由平面波基组展开,并由超软赝势实现离子实与价电子之间的相互作用势[10]; 结构弛豫使用BFGS算法[11],所有的计算都是在CASTEP软件包中完成[12].平面波截断能设置为470 eV,氮原子的2s22p3和钙原子的3s23p64s2轨道电子波函数被使用.布里渊区采用Monkhorst-Pack框架下的9×9×9 k点方案[13].这些设置可以使总能量的收敛标准达到0.01%以内.在几何优化中,体系总能量收敛标准小于每原子1.0 × 10-6 eV,作用力小于0.02 eV/nm.采用实验结构(a=b=c=0.772 1 nm)作为理论计算的初始构型[7].晶体结构中包含两种Ca—N键类型,氮原子处于6个钙原子构成的八面体中,而钙原子处于8个氮原子构成的十二面体中(图 1).理论计算晶体坐标和实验结果比较(表 1),计算结果和实验符合得很好,表明计算是精确的、可靠的.
-
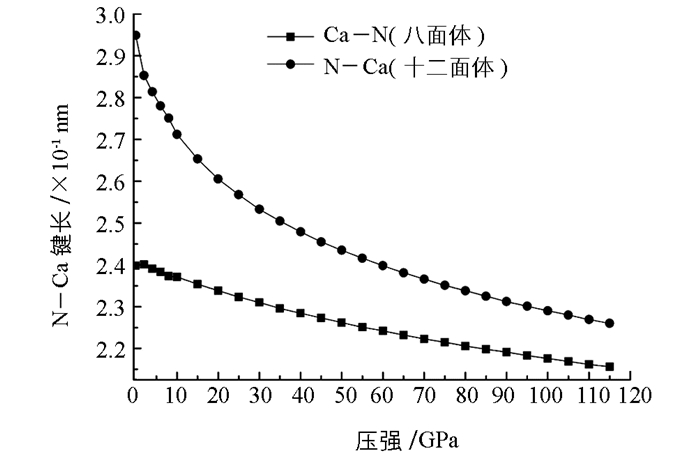
采用GGA交换关联势的BFGS方法研究了不同压强下γ-Ca3N2晶体的性质.为了确认晶体的稳定性和计算精度,表 2中给出了零压强下晶体的晶格参量、晶胞体积、键长和弹性模量,并与实验数据和其他理论结果进行比较.从表 2中可以发现,本文计算的晶格常数a与实验值的相对误差只有0.8%,晶胞体积误差为2.4%,十二面体中氮原子与钙原子之间的键长理论值与实验值的误差为2%,而八面体中误差只有1.5%.实验值中未给出γ-Ca3N2晶体的弹性模量,但是与其他理论计算的类似结构的弹性模量相近.这些结果进一步表明本文计算结果是可靠的. γ-Ca3N2晶体结构中两种不同类型的Ca—N化学键. 图 2给出了在不同压强作用下,这两种键长的变化情况.可以发现,在高的压强作用下,键长都被压缩了.同时,随着压强的增大,两种Ca—N键长之间的差别越来越小.十二面体中的Ca—N键长在0~50 GPa的压强范围内,变化相对比较陡峭,当压强增加到115 GPa时,键长减小了23%.而八面体中的Ca—N键长在压强作用下变化的比较缓慢,键长减小了10%.因此,这些结果表明八面体中的Ca—N键长比十二面体中的Ca—N键长有较小的压缩率.
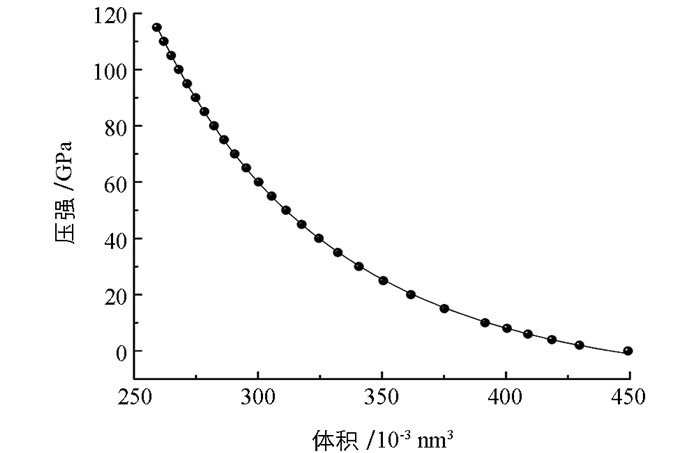
体弹模量B0作为表征材料硬度的一个参量,可以通过物态方程拟合获得.利用(1)式Birch-Murnaghan关系拟合压强-体积曲线(图 3). (1)式中x等于V/V0,拟合B0时,设定B′0等于4.43. 表 2中的体弹模量数值即为拟合结果.
对于立方晶体的力学稳定性判据,弹性系数Cij满足
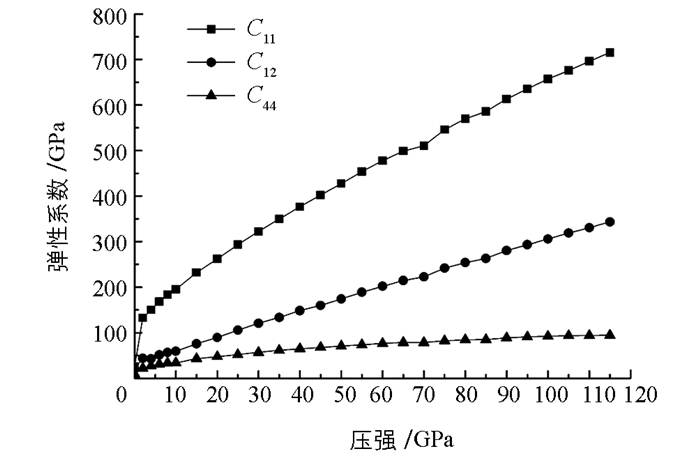
图 4给出了弹性系数Cij随压强的变化关系.从图 4中可以发现,在0~115 GPa的压强范围内,始终满足稳定性判据.同时,随着压强的增加,弹性系数也在增大.因此,对于立方结构的γ-Ca3N2晶体无论在低压下还是高压下都保持了力学稳定性.
-
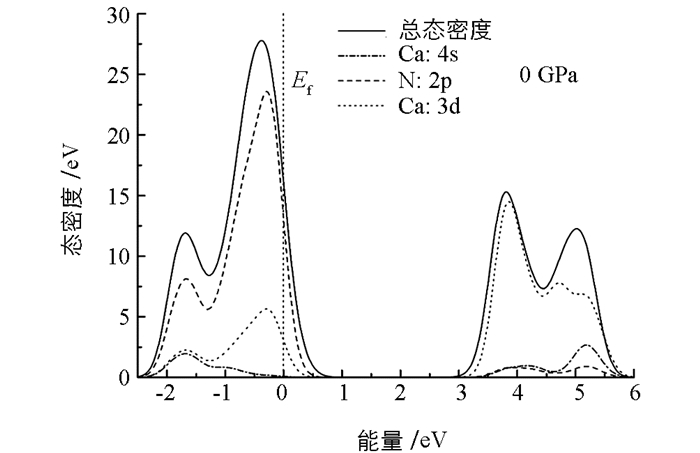
为了理解γ-Ca3N2晶体态密度(DOS)随压强的变化关系,首先分析了零压下的态密度(图 5).对于γ-Ca3N2晶体,零压下存在一个2.873 eV的宽直接带隙(导带底最低点和价带顶最高点之间的能量差值).费米能级被设置在能量为零的位置.在价带的上部,氮原子的2p电子对态密度贡献较多,2s轨道电子则较少.钙原子的电子结构比较特别,其3d空轨道也会影响自身的能带结构.非占有的钙原子3d态和4s电子都对价带有贡献,分别在-0.273和-1.715 eV能量位置出现两个峰.在能带结构中,导带也比较强.也可以发现由非占有的钙原子3d态和4s电子贡献的导带区域.同时,零压下N-2p和Ca-4s态密度在3.496 eV和4.735 eV能量附近存在轨道杂化.
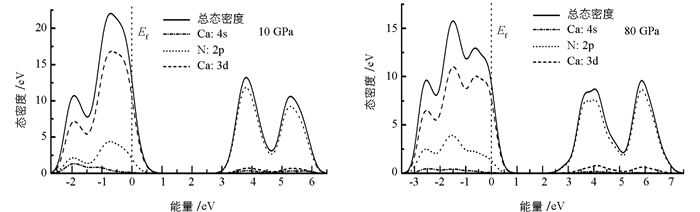
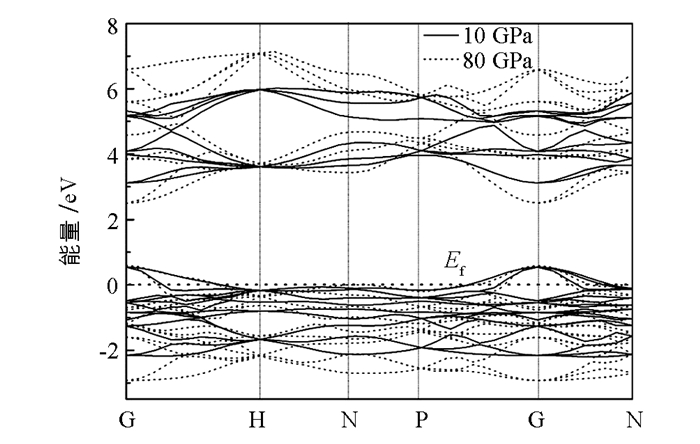
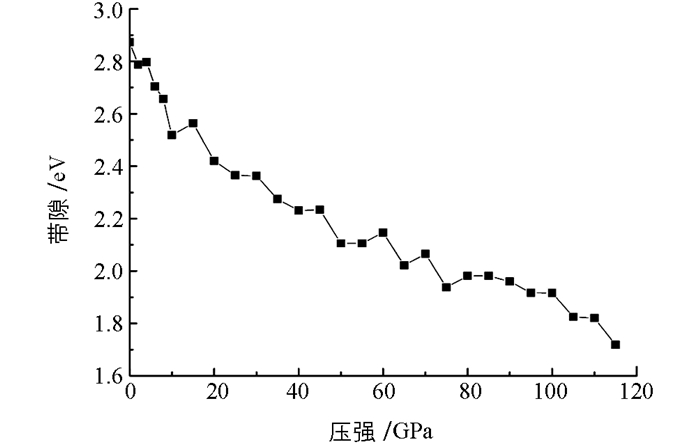
进一步研究高压下的态密度分布,使用主自旋态之和,分别计算了钙原子s,d轨道和氮原子的p轨道的态密度分布(图 6).比较图 5和图 6可以发现,随着压强的增大,态密度分布强度减小,导带底向低能区域移动.在80 GPa的压强下,价带峰发生了分裂,同时,N-2p和Ca-4s态的杂化程度,随压强增大而减小,说明化学键级也被改变. 图 7给出了10 GPa和80 GPa下的能带结构,从图 7中可以发现带隙变化比较明显,结果和图 6是一致的. 图 8为带隙随压强的变化关系,在高压下,材料的带隙明显减小.从图 5-7中,也可以看出,费米能级进入到价带,表明γ-Ca3N2晶体材料为p型半导体材料.
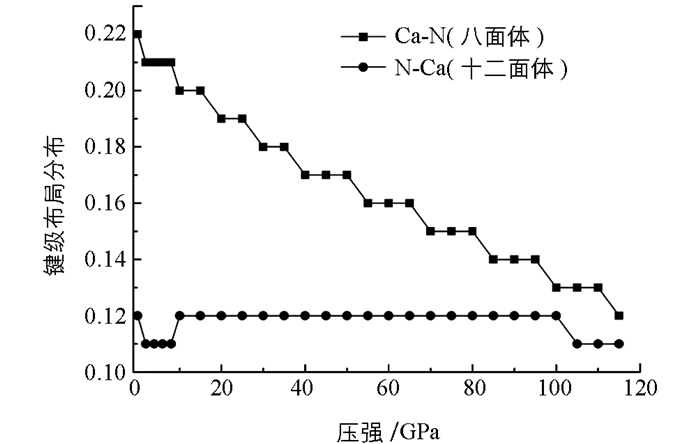
Mulliken布局分析常被用来讨论材料化学键的共价性和离子性[14].利用Mulliken方法计算了共价轨道的轨道占据和电荷分布. γ-Ca3N2晶体在不同压强下的离子组态见表 3.零压下的离子组态为Ca+0.83N-1.11,这里的“+”和“-”号分别代表失去和得到电荷.随着压强的增大,钙原子和氮原子之间有较小的电荷迁移.同时得到了不同压强下N—Ca键的键级布局分布(图 9).从图 9中可以发现,八面体和十二面体中的N—Ca键布局分布的变化有着显著的不同.八面体中的N—Ca键的变化陡峭,而十二面体中的N—Ca键的变化平缓.当压强增加时,这种键级布局分布的差别逐渐减小.这些结果说明γ-Ca3N2晶体中键的共价性随着压强增大而增强.需要指出的是Mulliken分析数值与原子轨道线性组合基组的选取有关,分析成键具有局限性,但是对电荷的变化趋势分析是可靠的.
-
知道电子波函数和能量,就可以计算出介电常数[15]ε(ω)=ε1(ω)+iε2(ω),介电常数的虚部ε2(ω)可以采用Kubo-Greenwood公式[16]从直接内部带迁移中获得.
其中,ħω为声子能量,Ω为晶胞体积,ψkn(r)和ψkl(r)分别为始末态波函数,fki和εki分别表示ith态的费米配分函数和电子能量.介电常数的实部ε1(ω),可以通过Kramers-Kronig(KK)关系获得[17]:
其中M表示积分的初始值.可以得出吸收系数α(ω)的表达式[18]为
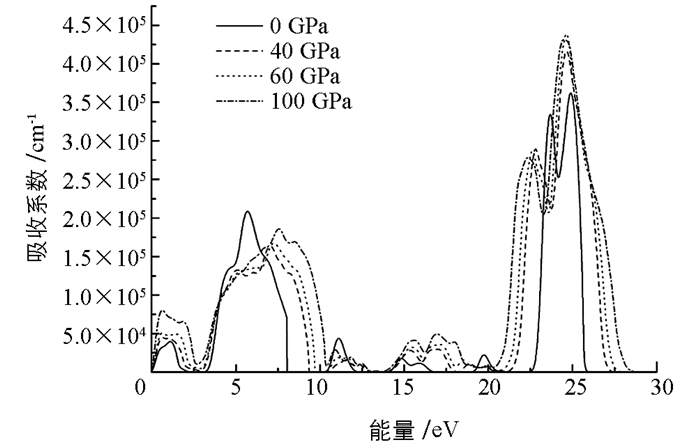
图 10给出了不同压强下γ-Ca3N2晶体的吸收系数.吸收光谱的性质决定于γ-Ca3N2晶体的结构,因为γ-Ca3N2晶体是各向同性的,所以光谱也表现出各向同性.计算所得光谱只包含直接内带跃迁.零压下光谱中主要有7个吸收峰,分别属于近红外光谱区、可见光谱区和紫外光谱区.高压下吸收光谱主要集中在紫外光谱区.随着压强的增大,紫外光谱区有向低能区移动的趋势.由于在压力的作用下,价带向高能区移动,而导带向低能区都扩展,致使电子跃迁需要吸收的能量减小,所以光谱有向低能(红移)方向移动的趋势.光谱带隙逐渐变窄,这一结果和直接带隙的变化趋势是一致的.这些结果表明Ca3N2晶体有可能作为高压下的光谱器件材料.
2.1. 几何结构和弹性特征
2.2. 电子结构和电荷分布
2.3. 光学性质
-
基于密度泛函理论的第一性平面波赝势方法,研究了0~115 GPa高压下立方Ca3N2晶体的几何结构,电子结构和光学性质.常压下的晶体结构参数很好的符合了实验结果.晶体中两种不同类型的Ca—N键长随压强的变化,说明八面体中的Ca—N键比十二面体中的Ca—N键有较小的压缩率.立方Ca3N2晶体拥有2.873 eV的宽带隙,随着压强的增大,带隙减小.基于Mulliken布局分析,发现随着压强的增加,Ca3N2晶体表现出较高的共价性.同时,吸收光谱在高的压力下表现出红移的趋势.本文的研究为更好地理解高压下Ca3N2晶体性质提供了参考.