


 下载:
下载:
-
慢性荨麻疹(Chronic urticaria,CU)是以反复发生的风团、瘙痒为主要表现的常见疾病,病程常迁延达6周以上. CU的病因复杂且隐匿,因此将大多数(>80%)无明显诱因的CU归为慢性自发性荨麻疹(Chronic spontaneous urticaria,CSU)[1]. 大量研究表明,CSU患者常同时伴随幽门螺杆菌(Helicobacter pylori,Hp)感染,经规范抗Hp治疗后,大部分患者的荨麻疹症状可得到缓解或完全消退,这些研究均提示Hp感染和CSU发病有着非常密切的相关性[2-5].
肥大细胞的激活是荨麻疹发病的关键,它常由其胞膜外的FcεRI受体和IgE交联触发[1]. 关于Hp相关体液免疫与CU发病机制的研究一直热度不断,Hizal等[6]发现血清抗Hp IgG阳性的荨麻疹患者自体血清皮肤试验阳性率(40%)显著高于血清抗Hp IgG阴性的荨麻疹患者(14.3%),而自体皮肤血清试验已被广泛用于检测与CSU发病密切相关的抗肥大细胞FcεRI抗体,Sun等[7]也发现抗Hp抗体阳性和抗TGAb阳性的CU患者血清抗FcεRI抗体阳性率较对照组显著增高. 但也有一些研究表示Hp感染与致CSU发病的自身抗体的产生无关[8]. Hp是一种能长期定植于胃肠道粘膜上皮细胞的微需氧革兰氏阴性杆菌,宿主持续的带菌状态可产生特异性体液免疫,导致感染局部粘膜和全身抗Hp特异性抗体高表达,包括IgA,IgE和IgG类抗体. 鉴于肥大细胞的胞膜外尚有多种正性效应的受体,除了对IgE表现高亲和力的FcεRI受体外,循环免疫复合物受体、低亲和力的IgG受体、补体受体等的特异性结合也可促使肥大细胞活化. Acuña等[9]对30例CU进行检测,其中血抗Hp IgG抗体阳性率为60.0%,IgM阳性率为33.3%;Galadari等[10]也发现血清抗Hp IgG,IgM抗体阳性率在CU患者中显著增高;Rostamy等[11]对43例CU患者血清特异性抗Hp-IgG,IgA,IgG联合IgA进行检测,发现阳性率分别为72.1%,46.5%和44.18%,与40例正常对照(37.5%,40%,27.5%)比较差异具有统计学意义;Liutu等[12]甚至用免疫印迹的方法在Hp中发现了IgE结合表位,提示Hp诱导的循环细菌特异性抗体可能在CU发病中起了重要作用. 更早期的研究对比均伴Hp感染的CU及非CU患者,前组可检出高滴度血清抗Hp-Lpp20蛋白的IgG,IgA抗体,这也是目前为止仅有的一项提出Hp具体蛋白成分特异性抗体参与CU发病的研究[13]. 但到目前为止,Hp介导的体液免疫和CU/CSU发病的相关研究样本数量较少,针对大样本的CSU血清抗Hp和其Lpp20蛋白抗体水平的研究鲜有报道,迫切需要在CSU大样本中调研Hp特异性抗体水平,并探讨其可能的致病机制. 本研究拟纳入较大样本的CSU患者及对照组,检测各组血清抗Hp和抗Hp-Lpp20的IgE、IgG和IgA水平,明确抗体分布情况,并探讨Hp感染促发的体液免疫在CSU发病中的角色及可能的机制.
全文HTML
-
CSU病例的血清样本:2014年7月-2015年1月在重庆大坪医院就诊并确诊为CSU的211例患者,其中112例女性(排除妊娠),99例男性,年龄为14-69周岁(38.79±4.22岁). 本研究纳入的CSU:①均符合2018年欧洲变态反应和临床免疫学(EAACI)会议的CSU诊断标准[1];②病程6周至7年不等,平均1.8年;③无用药史,近一周否认抗组胺治疗,近一月否认使用糖皮质激素、免疫抑制剂;④否认重大基础疾病及遗传、传染病病史;⑤从未接受过规范的抗Hp治疗. 健康对照组的血清样本:同期大坪医院体检中心常规体检结果正常的志愿者,对照组在年龄、性别等一般条件上与CSU组匹配. 纳入志愿者137例,其中73例女性(排除妊娠),64例男性,年龄为18-66周岁(38.29±3.37岁). 纳入的对照组:①无过敏性疾病病史;②一般检查无异常(二便、血常规,肝肾功);③无重大基础疾病及遗传、传染病病史;④从未接受过规范的抗Hp治疗. EDTA抗凝管收集各受试者外周静脉血,2 h内静置离心、收集血清. 菌株和质粒Hp 26695标准株购自美国菌种保藏中心(ATCC);BL21(DE3) 感受态大肠杆菌购自北京天根公司;由本实验室制备的pET22b-Lpp20重组质粒已转入DH5α感受态大肠杆菌,氨苄西林(Amp)抗性[14].
-
标准Hp-ELISA血清检测试剂盒购于北京贝尔生物公司;质粒抽提试剂盒购于Omega公司;细菌基因组DNA抽提试剂盒购于北京达科为公司;Chelating Sepharose Fast Flow纯化柱购于Pharmacia公司;生物素-山羊抗人IgE,IgA购自英国AbD serotec公司;HRP-羊抗人IgG购自北京中杉公司;HRP-链霉亲和素购自美国Abbkine公司;TMB显色液购于北京天根公司;BCA蛋白浓度试剂盒购于Sigma公司;ECL化学发光试剂盒购于上海碧云天公司;N-端测序送中国科学院测定.
-
用标准的Hp-ELISA试剂盒就CSU组和对照组Hp感染阳性率(抗CagA及HSP联合抗原的IgG抗体)进行检测. 具体操作严格遵守说明书,酶标仪450 nm读取OD值.
-
(1) Hp的培养和菌体尿素裂解液的制备
① 细菌培养[14]:将保种的Hp菌液接种于固体Hp培养基上37 ℃培养长出的菌落,再次接种入布氏肉汤培养基(含10%胎牛血清)扩增到对数分裂期. ② Hp尿素裂解液的制备:菌液用磷酸盐缓冲盐溶液(PBS)重悬洗涤3次,离心后将菌体沉淀加入尿素溶液(8 M)中,充分吹打后即成,BCA法测蛋白质量浓度.
(2) 抗Hp-IgG,IgE和IgA抗体水平测定
① 间接ELISA法测定抗Hp-IgG:20 μg/mL的Hp尿素裂解液包被酶标板,加入1∶40稀释血清100 μL/孔,37 ℃孵育1 h,洗板后加入1∶3 000稀释的HRP-羊抗人IgG 100 μL/孔,孵育1 h,洗板后拍干板中液体,加入显色液避光显色15 min,终止反应,450 nm读取OD值. 每样本设3复孔,同时设无血清对照孔. ②生物素-亲和素ELISA法测定抗Hp-IgE/IgA:将各血清1∶20稀释后加样入Hp尿素裂解液包被的酶标板,37 ℃,1 h孵育后洗板,加入1∶10 000的生物素-山羊抗人IgE或1∶2 000的生物素-山羊抗人IgA孵育1 h,洗板,加入1∶6 000 HRP-链霉亲和素孵育35 min,加入TMB显色液显色20 min,终止反应,450 nm读取光密度(OD)值. 同时设置复孔及无血清对照.
-
(1) Lpp20的表达和纯化[14]
将pET22b-Lpp20-DH5α菌液培养扩增后提取重组质粒DNA并经NdeI+XhoI双酶切鉴定正确,再将质粒转入感受态BL21(DE3)中,构建工程菌pET22b-Lpp20-BL21(DE3)并诱导质粒表达. SDS-PAGE凝胶电泳法鉴定重组Lpp20蛋白的表达,镍离子亲和层析法纯化蛋白,蛋白浓度测定(BCA)法测定浓度,并送中国科学院进行N-端测序,增强化学发光法鉴定Lpp20重组蛋白的抗原反应性.
(2) 抗Lpp20-IgG,IgE,IgA抗体水平测定
取Lpp20以5 μg/mL的质量浓度,将各血清1∶20稀释加入包被好的酶标版中,检测各样本中抗Lpp20-IgG,IgE,IgA抗体水平. 同时设置复孔及无血清对照.
-
使用GraphPad Prism 5.0统计软件进行数据分析并作图,Hp感染的阳性率(%)行卡方检验比较,p小于0.05判定为差异具有统计学意义;正态计量资料用均数±标准差(x±s)表示,组间进行非配对t检验比较,p小于0.05判定为差异具有统计学意义.
1.1. 材料
1.1.1. 血清样本、菌株、质粒
1.1.2. 主要试剂
1.2. 试验方法
1.2.1. Hp感染阳性率测定
1.2.2. 血清特异性抗Hp全菌抗原IgG,IgE和IgA抗体检测
1.2.3. 血清特异抗Hp-Lpp20-IgG、IgE和IgA抗体检测
1.3. 统计学方法
-
CSU组和对照组均进行血清抗Hp-ELISA标准试剂盒检测,结果表明,在CSU组中,抗Hp抗体阳性率为73.46%(155/211),对照组中阳性率为51.82%(71/137),两组行卡方检验(χ2=17.08,p=0.000)具有统计学意义(表 1),可认为CSU发病与Hp感染有密切的相关性.
-
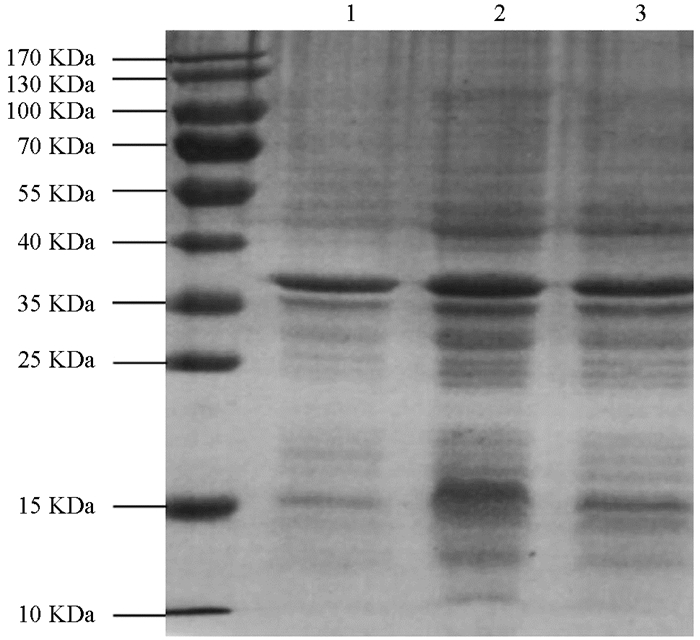
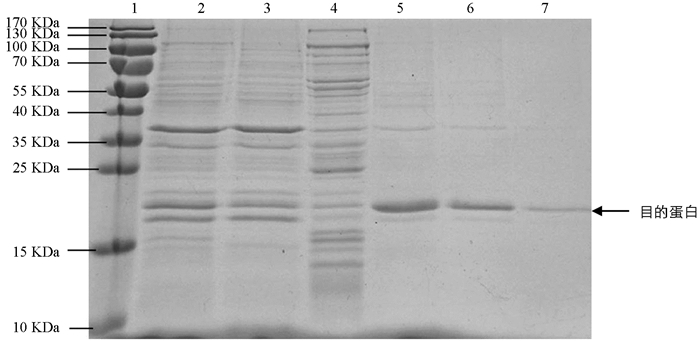
将诱导表达的pET22b-Lpp20-BL21用SDS-PAGE凝胶蛋白电泳进行检测,诱导4 h的工程菌在20 kDa附近有新的蛋白表达条带,目的蛋白表达成功(图 1). 目的蛋白的纯化使用镍离子亲和层析法,将不同设置参数下的收集液分别用SDS-PAGE电泳进行检测,其中目的蛋白纯度可达90%,经分子筛浓缩后BCA法测定蛋白质量浓度为2.2 mg/mL(图 2). 重组蛋白的N端测序为NH2-Met-Lys-Asn-Gln-Val,Fasta将此序列与蛋白质数据库进行比较,与预期的Lpp20的N-端蛋白序列一致;重组Lpp20蛋白经增强化学发光法鉴定具有良好的抗原反应性(图略).
-
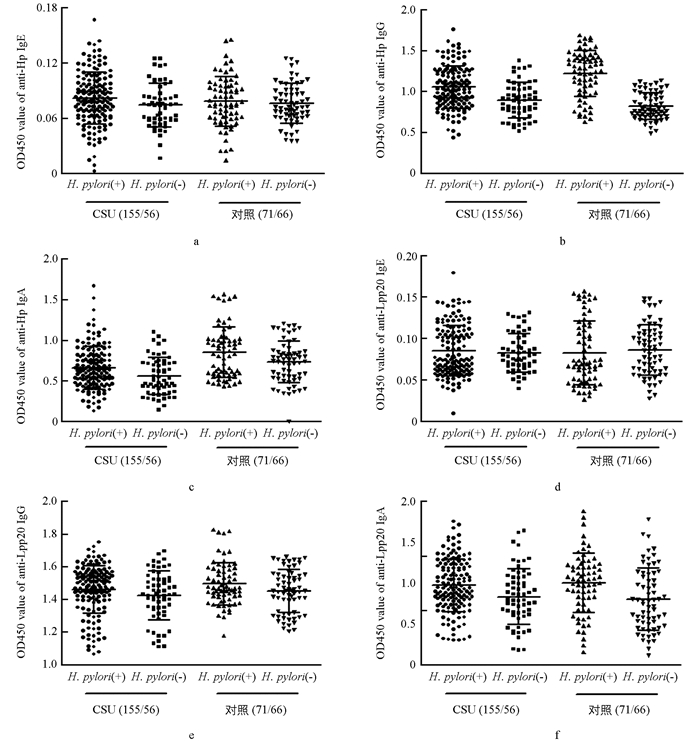
通过2.1的结果,将CSU组分为:①CSU(+)Hp(+)组,计155例,②CSU(+)Hp(-)组,计56例;将对照组分为:③CSU(-)Hp(+)组,计71例,④CSU(-)Hp(-)组,计66例. 经生物素-亲和素ELISA法测定显示,抗Hp-及抗Lpp20-IgE分别在①,②组间(0.082±0.002 vs 0.074±0.003,p1=0.076;0.085±0.003 vs 0.083±0.003,p2=0.622);①,③组间(0.082±0.002 vs 0.078±0.003,p1=0.382;0.085±0.003 vs 0.082±0.005,p2=0.597)比较,差异不具有统计学意义(图 3a、图 3d).
经间接ELISA法测定显示,抗Hp-IgG在①,②组间(1.053±0.022 vs 0.890±0.028,p=0.000),③,④组间(1.222±0.034 vs 0.816±0.020,p=0.000)比较,差异具有统计学意义,表明Hp感染后,人群中血清抗Hp全菌-IgG水平显著升高;①,③组间(1.053±0.022 vs 1.222±0.034,p=0.000)比较,①组抗体水平明显低于③组,且差异具有统计学意义(图 3b). 抗Lpp20-IgG在①,②组间(1.459±0.012 vs 1.422±0.019,p=0.10),①,③组间(1.459±0.012 vs 1.494±0.016,p=0.087)比较,差异不具有统计学意义;但③,④组间(1.494±0.016 vs 1.449±0.016,p=0.046)比较,差异具有统计学意义(图 3e). 经生物素-亲和素ELISA法测定显示,抗Hp-IgA在①,②组间(0.660±0.02 vs 0.559±0.030,p=0.01),③,④组间(0.849±0.037 vs 0.730±0.032,p=0.000)比较,差异具有统计学意义,表明Hp感染后,人群中血清抗Hp全菌-IgA水平显著升高;①,③组间(0.660±0.02 vs 0.849±0.037,p=0.019)比较,①组水平低于③组,且差异具有统计学意义(图 3c). 抗Lpp20-IgA在①,②组间(1.229±0.020 vs 1.124 ±0.033,p=0.005),③,④组间(1.251±0.033 vs 1.098±0.035,p=0.002)比较,差异具有统计学意义,表明Hp感染后人群中血清抗Hp-Lpp20-IgA水平显著升高;但是①,③组间(1.229±0.020 vs 1.251±0.033,p=0548)比较,差异不具有统计学意义(图 3f).
2.1. CSU组和对照组Hp感染阳性率
2.2. Lpp20重组蛋白的表达、纯化和鉴定
2.3. CSU组及对照组的血清抗Hp全菌抗原和抗Lpp20的IgE,IgG及IgA水平
-
越来越多的研究揭示了CSU发病与Hp感染相关[2-5],在EAACI最新发布的荨麻疹诊疗指南中,已将检测Hp感染情况纳入CSU常规诊疗计划中[1]. 本研究首先用标准化Hp-ELISA试剂盒检测了CSU人群和健康对照中Hp感染阳性率(包括既往感染和现症感染),数据显示CSU人群中Hp感染阳性率明显高于健康对照,证明Hp感染和CSU发病存在密切的相关性(表 1). 荨麻疹发生的关键病理机制是肥大细胞活化、脱颗粒,继而合成和释放炎性介质,而其中最经典的肥大细胞激活途径是IgE与其胞膜外的FcεRI受体病理性结合,Hizal等[6]发现血清抗Hp IgG阳性的荨麻疹患者自体血清皮肤试验阳性率(40%)显著高于血清抗Hp IgG阴性的荨麻疹患者(14.3%),而自体皮肤血清试验已被广泛用于检测与CSU发病密切相关的抗肥大细胞FcεRI抗体,Sun等[7]也发现抗Hp抗体阳性和抗TGAb阳性的CU患者血清抗FcεRI抗体阳性率较对照组显著增高,提示Hp感染可能通过诱导产生外周血抗FcεRI抗体升高的途径促进荨麻疹发生;但也有一些研究表示Hp感染和致CSU发病的自身抗体的产生无关. 同时,肥大细胞表面仍分布有其它正性调节受体,如循环免疫复合物受体、IgG受体等,这些受体在特异性体液免疫亢进时和相应的配体结合,也能够引起细胞活化,促进后继炎症反应[1]. 感染Hp后的长期带菌状态可使宿主产生持续性体液免疫,导致感染局部粘膜和循环抗Hp抗体水平增加,包括IgG,IgA和IgE等,虽然这些抗体对于清除Hp定植作用甚微,却可能在其他病理机制中发挥作用. 如已有研究揭示,在伴随Hp感染的CU病例中,血清抗Hp-IgG,IgA水平显著高于伴随Hp感染的非CU对照. Bakos等[13]的研究也发现,在伴随Hp感染的CU受试组中,存在高滴度血清抗Hp-Lpp20-IgG及IgA,这些研究都提示Hp感染介导的体液免疫可能在某些CU发病中发挥了重要作用. 但既往相关研究纳入的样本量极少,迄今未见关于大样本CSU中Hp全菌及Lpp20血清特异性抗体的调研分析,因此本研究就CSU患者和健康对照中抗Hp全菌和其Lpp20血清特异性的IgE,IgG和IgA水平进行调研,以期揭示Hp引起的体液免疫在CSU发病中所扮演的角色.
本研究通过对受试血清样本细菌特异性IgE进行检测发现,在各受试组中(CSU(+)Hp(+)组、CSU(-)Hp(+)组、CSU(+)Hp(-)组、CSU(-)Hp(-)组),血清特异性抗Hp全菌-和Lpp20-IgE抗体均处于差异不具有统计学意义的低值水平(组间比较p均大于0.05)(图 3a、图 3d),表明Hp感染不能有效激发机体的IgE型体液免疫,且所产生的低水平细菌特异性IgE与CSU发病无相关性. 通过检测受试血清中细菌特异性IgG发现,不论在CSU组还是对照组,感染Hp后均能引起外周循环中抗Hp全菌-IgG显著升高(p < 0.05),但CSU(+)Hp(+)组中抗Hp全菌-IgG水平显著低于CSU(-)Hp(+)组(p < 0.001,图 3b),揭示在CSU人群中,Hp感染后激发IgG型体液免疫的强度远远小于无CSU人群,我们因此推测虽然感染Hp可诱导宿主产生高水平特异性的IgG抗体,但是这种体液免疫效应并不是促进CSU发病的重要病理机制. 同样的,不论在CSU组还是对照组中,感染Hp后血清中抗Lpp20-IgG水平均升高,但这种升高趋势只在CSU(-)Hp(+)组中差异具有统计学意义,并且CSU(+)Hp(+)组血清抗Lpp20-IgG水平也低于CSU(-)Hp(+)组,但两组间差异不具有统计学意义(图 3e),因此我们认为Hp感染可以通过其Lpp20蛋白诱导宿主产生较高水平的血清特异性IgG,虽然Lpp20是Hp中具备良好免疫原性的成分蛋白,但它却不是Hp感染致CSU发病的重要病因. 通过检测受试血清细菌特异性IgA发现,不论在CSU组或对照组,感染Hp后都会引起血清抗Hp全菌-和抗Lpp20-IgA显著升高(p < 0.05),且CSU(+)Hp(+)组中血清特异性抗Hp全菌-IgA水平显著低于CSU(-)Hp(+)组(p < 0.05),而这两组间的抗Lpp20-IgA水平差异不具有统计学意义(图 3c、图 3f). 我们推测,虽然感染Hp可激发宿主产生血清高水平特异性IgA,而Hp-Lpp20又是参与此反应的具备良好免疫原性的成分,但是这种IgA型体液免疫反应并非Hp感染相关性CSU的主要致病机制. 至于CSU(+)Hp(+)组中抗Hp全菌-IgG,IgA水平较CSU(-)Hp(+)组更低(p < 0.05),我们推测是由于部分病例Hp感染后更向Th1型免疫反应偏移[15],表现为血清抗体水平低,而胃肠道粘膜细胞免疫增强使粘膜损伤更严重,细菌或其代谢产物可直接接触胃肠道固有层肥大细胞,进一步使其活化而诱发包括荨麻疹在内的炎症反应;或者也可能是CSU宿主在荨麻疹炎症状态和Hp感染定植的相互作用下,能够通过某些机制削弱Hp介导的体液免疫反应. 已有研究表明,CU患者血清sIL-2R水平与类胰蛋白酶水平显著相关,提示CU患者T细胞活化与肥大细胞脱颗粒成正比,CU患者具有更亢进的外周血T细胞活性[16].
我们有理由推断,虽然感染Hp可以导致宿主产生高水平的血清特异性IgG,IgA,并且Hp-Lpp20因具备良好的免疫原性;也在Hp感染触发的体液免疫中占有重要权重,但在与Hp感染相关的CSU病程中,Hp细菌特异性抗体的产生并不是主要致病机制,也即Hp并不是通过诱发体液免疫促进CSU发病. 我们认为,机体感染Hp后存在除体液免疫外的其他一些途径促进肥大细胞活化参与CSU发病. 已经有一些研究表明,某些Hp蛋白如VacA和NAP,可通过引起肥大细胞的胞内钙振荡或G蛋白介导的MAPK,PI3K/Akt信号通路活化的方式直接激活人肥大细胞[17-18],提示Hp感染后极有可能通过直接作用的方式活化粘膜固有层肥大细胞,促进CSU发病. 我们的研究成果也更加全面地对Hp这种直接活化肥大细胞的方式做了补充[19].
本研究揭示了Hp感染和CSU发病的相关性,首次在大样本的CSU病例中调研了血清特异性抗Hp全菌-及Lpp20-IgE,IgG,IgA水平,论证了Hp血清特异性抗体并非Hp感染相关性CSU的病因. 虽然我们的研究并未完全揭示Hp感染致CSU的发病机制,但却给相关研究提供了新的思路,避免了某些研究资源的浪费.