


 下载:
下载:
-
开放科学(资源服务)标识码(OSID):

-
灰葡萄孢菌(Botrytis cinerea)是一种典型的死体营养型病原菌,引起的灰霉病是一种具有毁灭性的真菌病害,其产孢量大、寄主范围广泛,植物整个生育期皆可发病,低温高湿条件下灰霉病加重,可引起作物腐烂或幼苗死亡,严重威胁果蔬等重要经济作物的产量和质量[1-2]。
目前防治灰霉病主要以化学防治为主,常用的杀菌剂有苯并咪唑类、二甲酰亚胺类、苯胺基嘧啶类、苯吡咯类、琥珀酸脱氢酶抑制剂类等[3-4]。由于灰葡萄孢菌具有寄主范围广、繁殖速度快、遗传变异强以及寄生适合度高等特点,导致其在过去几十年大量用药情况下对多种作用机制的杀菌剂均产生了严重的抗性,增加了抗药性的风险[5]。
1967年由日本公司研究发现二甲酰亚胺类杀菌剂(Dicarboximide Fungicides,DCFs)对灰葡萄孢菌具有良好的防治效果[6],并在20世纪70年代首次引入中国市场[7],主要包括异菌脲、腐霉利和乙烯菌核利等。但随着该类药剂的广泛使用,灰葡萄孢菌对其抗性问题日益严重。据报道,1979年德国和法国分别有近80%和60%的灰葡萄孢菌对该类药剂产生抗性;2021年,巴西灰葡萄孢菌对异菌脲的抗性达到44%[8]。在中国各地都检测到了高频的DCFs抗性菌株[9-10],2013年宋晰等[9]研究表明在内蒙古、辽宁等地采集的灰葡萄孢菌对腐霉利的平均抗性频率均高达90%以上;2018年,杜颖[10]发现辽宁对腐霉利抗性达到94%以上;2021年,四川灰葡萄孢菌对腐霉利抗性频率达80%[11];2022年,江苏灰葡萄孢菌对异菌脲抗性频率达51.44%[12]。
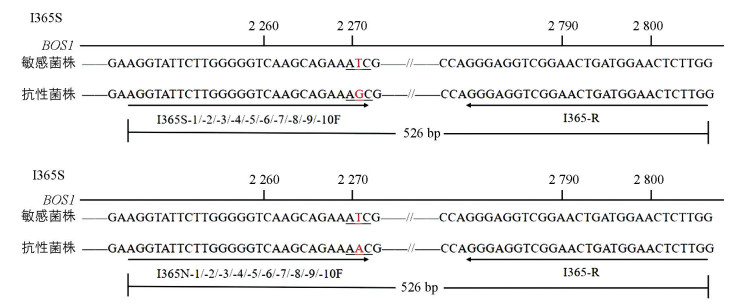
该类药剂抗性机理可能是通过过度激活相关信号的传导途径造成甘油等渗透压稳定剂在菌体内大量积累,从而导致外部水的大量涌入造成细胞膨胀后死亡[13]。植物病原真菌抗药性的产生大多是由特定基因的单核苷酸突变导致[14-15]。Orth等[16]认为DCFs抗性是由单基因控制的,其靶标位点可能与双组份组氨酸激酶(TCHK)有关,真菌中双组分组氨酸激酶通常由5~6个重复HAMP区域、HisKA、HATPase、REC等结构域组成,但该类药剂与靶标位点的精确作用机理尚不明确[17-19]。灰葡萄孢菌双组分组氨酸激酶信号途径中包括双组分组氨酸激酶BOS1、反应调控蛋白BRRG-1、促分裂原活化蛋白激酶(Mitogen-activated-ProteinKinase,MAPK)BOS2和MAPK的激酶BOS4、BOS5等重要元件,但仅双组分组氨酸激酶BOS1对DCFs抗性起作用[20]。目前,很多研究者认为双组分组氨酸激酶BOS1基因保守区域HAMP点突变导致灰葡萄孢对该类杀菌剂抗性的产生。通过对比不同国家中采集的灰葡萄孢菌DCFs抗性和敏感菌株的DNA序列,发现抗性菌株中分别存在BOS1基因365位或369位氨基酸替换[21-22];Banno等[23]和Ma等[24]研究者也表明BOS1上I365SV、V368F、Q360P和Q396H点突变与DCFs抗性有关。除该基因点突变外,其他抗性机制也可能会导致抗药性产生(如链格孢菌[25]),但这种机制目前还未在灰葡萄孢菌中有相关报道。现已报道的BOS1上存在的点突变有I365N/R/S、Q369P/H、V368F、N373S和T447S等,其中365、369、373位氨基酸的突变最为常见,这些突变会使HAMP区域的空间构象发生改变从而导致灰霉病菌对DCFs产生不同水平的抗性[26-27]。研究结果显示,重庆地区灰葡萄孢菌对DCFs抗性BOS1I365S、BOS1I365N点突变占主导地位,比例分别为37.5%和52.1%[28]。
及时了解和发现地区抗药性发展情况,可根据结果更改用药策略,因此掌握病原菌抗药性的快速检测方法也显得尤为重要。检测杀菌剂抗性的传统方法包括菌落直径法、孢子萌发法、抑菌圈法和最低浓度抑制法等[29-34],但此类方法检测周期长、操作繁琐、效率低。分子检测方法具有简单、快速和通量高等优点。等位基因特异性PCR(Amplification Refractory Mutation System PCR,AS-PCR)就是一种高通量检测单核苷酸突变的分子技术,该方法操作简单、特异性强、成本相对较低,只需要PCR仪和琼脂凝胶电泳即可完成检测。如Ma等[35]、罗梅等[36]和郭东锋等[37]利用AS-PCR技术分别成功检测了白僵菌对苯并咪唑类杀菌剂、桃褐腐病菌对多菌灵以及马铃薯晚疫病菌对甲霜灵的抗性菌株。本研究基于灰葡萄孢菌DCFs抗性突变靶标BOS1基因I365S和I365N点突变,建立了能快速检测抗性菌株的AS-CR技术并将其初步应用于田间抗性监测,旨在实践中对灰葡萄孢菌DCFs抗性群体进行动态监测。
全文HTML
-
灰葡萄孢菌BOS1I365S点突变菌株CS3、CS8、H9和XM9,BOS1I365N点突变菌株B1、B31、B32和XM5,敏感菌株B05.10、C25、lp1和H4;小麦赤霉病菌、高粱炭疽病菌、稻瘟病菌、稻曲病菌、核盘菌和木霉菌。
-
从NCBI网站下载二甲酰亚胺类杀菌剂作用靶标双组分组氨酸激酶的编码基因BOS1(序列号:AF396827.2),并根据抗性菌株BOS1基因HAMP区域点突变I365S和I365N(突变位置见图 1),同时借助于软件primer premier 5.0完成引物设计,对相应的突变方式分别设计相应的正向特异性引物,将突变位点设计在正向引物的3'末端,且在突变点前后1~2个碱基设计错配进而提高引物的特异性,引物序列如表 1、表 2。以CTAB法[38]提取的灰葡萄孢敏感菌株和抗性菌株DNA为模板进行PCR扩增,扩增PCR产物预期大小为526 bp。
-
为明确10对引物是否可以区分敏感菌株和抗性菌株,分别以敏感菌株B05.10、BOS1I365S点突变菌株CS3,BOS1I365N点突变菌株B1的DNA为模板进行AS-PCR进行扩增,ddH2O作为空白对照。每组引物设置8个退火温度(68 ℃、67 ℃、66 ℃、63 ℃、61 ℃、58 ℃、57 ℃和56 ℃)进行扩增。反应体系:2×Taq Master Mix 12.5 μL、10 μmol/L正反向引物各1 μL、DNA模板1 μL和ddH2O补足至25 μL;反应程序:95 ℃预变性3 min;95 ℃变性30 s,退火30 s,72 ℃延伸1 min,共34次循环;72 ℃终延伸10 min。PCR反应后用1%琼脂凝胶电泳并记录结果。
-
为避免假阴性出现,本实验特引入内参引物进行多重AS-PCR扩增。内参引物选用真菌的通用ITS引物ITS4(5′-TCCTCCGCTTATTGATATGC-3′)和ITS86(5′-GTGAATCATCGAATCTTTGAAC-3′)。内参引物和特异性引物的浓度比例分别设置为1∶1、1∶2、1∶4和1∶5,即特异性引物浓度为10 μmol/L,内参引物浓度分别为10 μmol/L、5 μmol/L、2.5 μmol/L和2 μmol/L。反应体系同1.2.2,程序中退火温度为最适退火温度。
-
为评价引物的特异性,分别选择小麦赤霉病菌、高粱炭疽病菌、稻瘟病菌、稻曲病菌、核盘菌、木霉菌以及灰葡萄孢菌标准菌株B05.10和抗性菌株(CS3和B1)DNA作为模板,反应体系和程序同1.2.3。
-
将提取的抗性菌株DNA模板梯度稀释为原浓度的100、10-1、10-2、10-3、10-4、10-5和10-6。得到7个DNA浓度梯度进行AS-PCR反应灵敏度检测,反应体系和程序同1.2.3。
-
本研究采用灰葡萄孢菌菌株进行检测体系的验证,即BOS1I365S(CS8、H9和XM9)和BOS1I365N(B31、B32和XM5)点突变菌株和敏感菌株(C25、lp1和H4)的DNA为模板,反应体系和程序同1.2.3。
-
本研究利用在重庆市北碚区本勋农业园(106.44°E,29.76°N)采集的草莓灰霉病病果,挑取病果表面的菌丝和孢子,采用NaOH裂解法[39]直接快速提取病原菌DNA,反应体系中DNA量相应改为5 μL,其他成分和反应程序同1.2.3。将AS-PCR检测结果为阳性的部分菌株PCR扩增的BOS1基因产物送至上海生物工程有限公司测序,将测序结果翻译成蛋白序列与NCBI中标准菌株B05.10用DNAMAN软件进行氨基酸比对。
1.1. 供试菌株
1.2. 试验方法
1.2.1. AS-PCR引物设计
1.2.2. AS-PCR反应退火温度的确定
1.2.3. 多重AS-PCR特异性引物与内参引物浓度的确定
1.2.4. 多重AS-PCR反应体系特异性的检测
1.2.5. 多重AS-PCR反应体系灵敏度的检测
1.2.6. AS-PCR检测体系的验证
1.2.7. AS-PCR检测体系田间应用
-
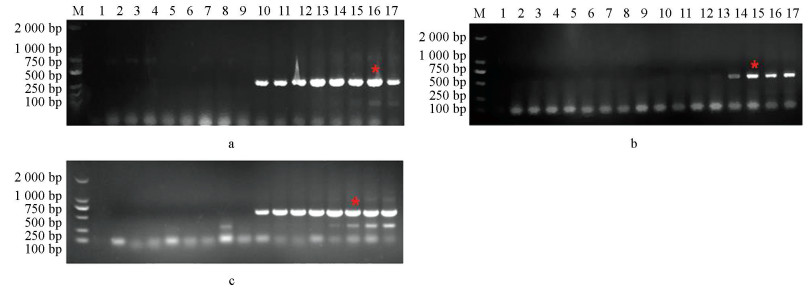
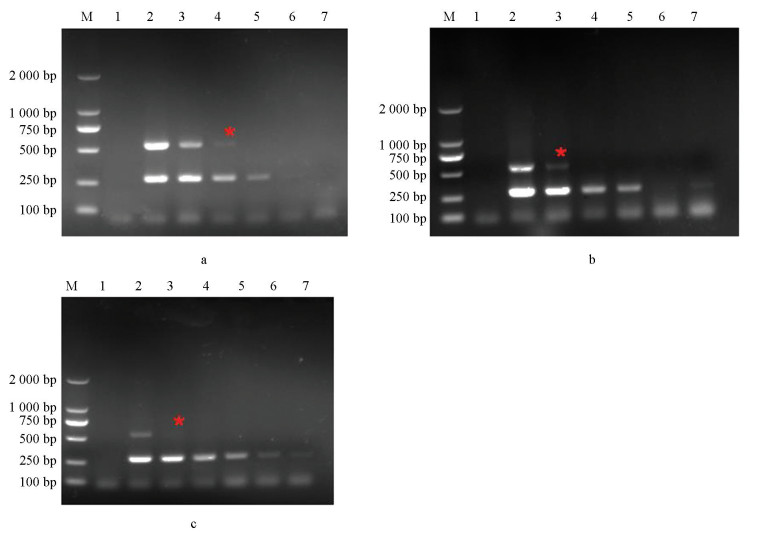
利用上述根据突变位点设计的引物,分别以BOS1I365S点突变菌株CS3、BOS1I365N点突变菌株B1和敏感菌株B05.10的DNA为模板进行PCR扩增,引物对I365-7F/I365-R和I365S-8F/I365-R均只能在以BOS1I365S点突变菌株为模板情况下扩增出条带(图 2a、2b);引物对I365N-8F/I365-R也仅在以BOS1I365N点突变菌株为模板情况下才能扩增出条带(图 2c)。说明筛选过的这3组引物可区分敏感和抗性菌株,由此,选择这3组引物进行后续试验。
-
以BOS1I365S点突变菌株CS3、BOS1I365N点突变菌株B1和敏感菌株B05.10为模板,在不同退火温度下进行扩增,电泳检测结果如图 2。引物对I365S-7F/I365-R和I365N-8F/I365-R在退火温度为56~68 ℃时均能扩增出条带(图 2a、2c),引物对I365S-8F/I365-R在退火温度为56~61 ℃时扩增出条带(图 2b),且图 2a中16泳道、b和c中15泳道条带最亮,最终确定引物对I365S-7F/I365-R退火温度为57 ℃、I365S-8F/I365-R和I365N-8F/I365-R退火温度为58 ℃。
-
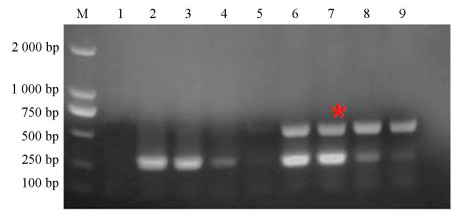
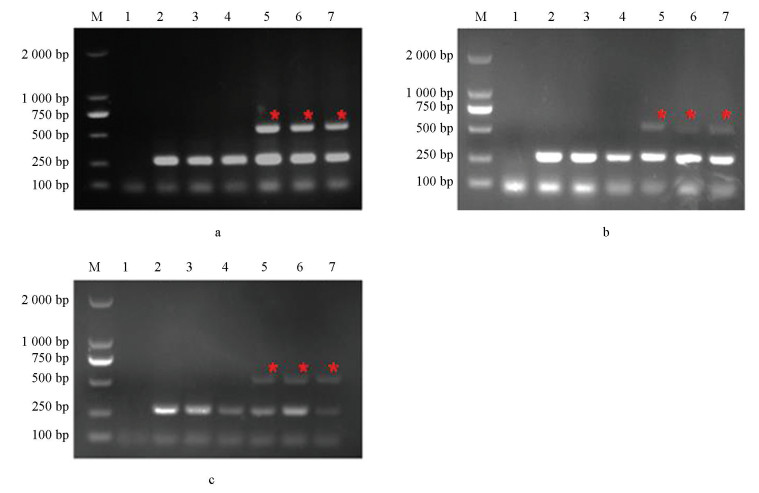
真菌通用引物为ITS1/ITS4,但由于该组引物扩增出的内参片段大小与目标片段差距不大,在利用凝胶电泳进行检测时无法区分目标片段与内参片段。所以本研究选用ITS4/ITS86作为内参引物,并利用该组通用引物与特异性引物进行多重AS-PCR扩增。当特异性引物与内参引物浓度比例为4∶1时,敏感菌株扩增出的内参条带较微弱,但当比例为2∶1时,敏感和抗性菌株内参条带扩增条带较亮且更加稳定(图 3)。因此,最终确定内参引物与特异性引物浓度比例为1∶2,即内参引物浓度为5 μmol/L。
-
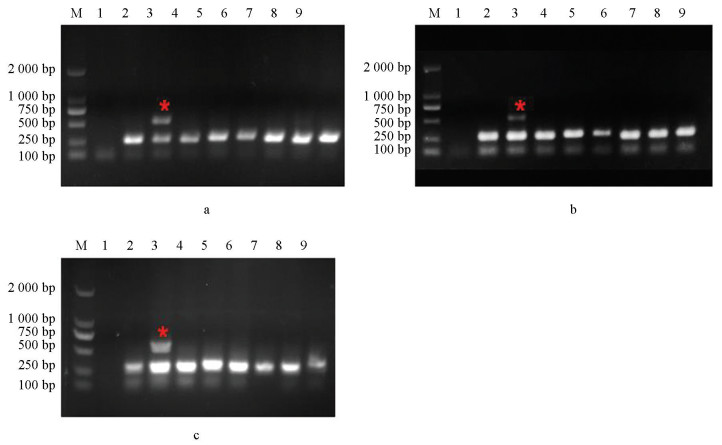
本研究选用不同真菌以及灰葡萄孢标准菌株B05.10和抗性菌株(CS3和B1)对该体系的特异性进行评价,分别以该菌株为模板进行PCR扩增,结果如图 4所示,其a、b和c分别对应引物对I365S-7F/I365-R、I365S-8F/I365-R和I365N-8F/I365-R,只有抗性菌株能够扩增出特异性和内参2种条带,其他真菌均只能扩增出内参条带,说明该体系具有特异性。
-
将抗性菌株模板(70 ng/μL)梯度稀释后进行扩增,电泳结果见图 5,a中引物对I365S-7F/I365-R可在稀释度10-2时扩增出特异性条带,b和c中引物对I365S-8F/I365-R和I365N-8F/I365-R可在稀释度为10-1时扩增出特异性条带,即引物对I365S-7F/I365-R、I365S-8F/I365-R和I365N-8F/I365-R的灵敏度分别为0.7 ng/μL、7 ng/μL和7 ng/μL。
-
本研究首先选用3株敏感菌株(C25、lp1和H4)和6株分别为I365S(CS8、H9和XM9)、I365N(B31、B32和XM5)突变方式的抗性菌株来检测这4组引物特异性稳定性。如图 6所示,a、b和c分别对应引物对I365S-7F/I365-R、I365S-8F/I365-R和I365N-8F/I365-R,3组引物在以抗性菌株为模板的情况下均能扩增出2条带,在以敏感菌株为模板的情况下只能扩增出1条带,说明该体系能够区分抗性和敏感菌株。
-
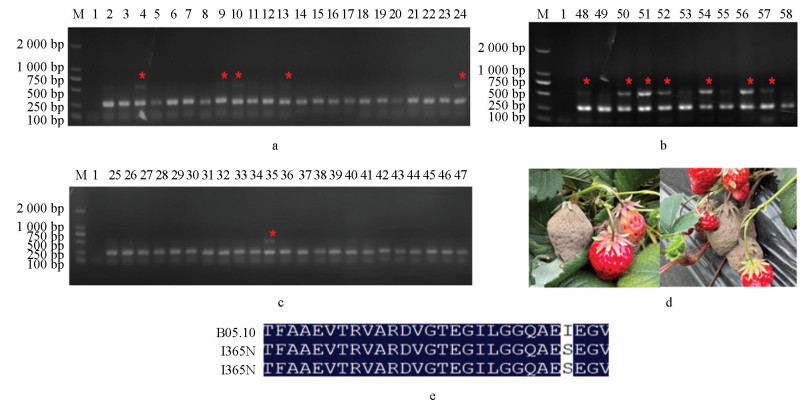
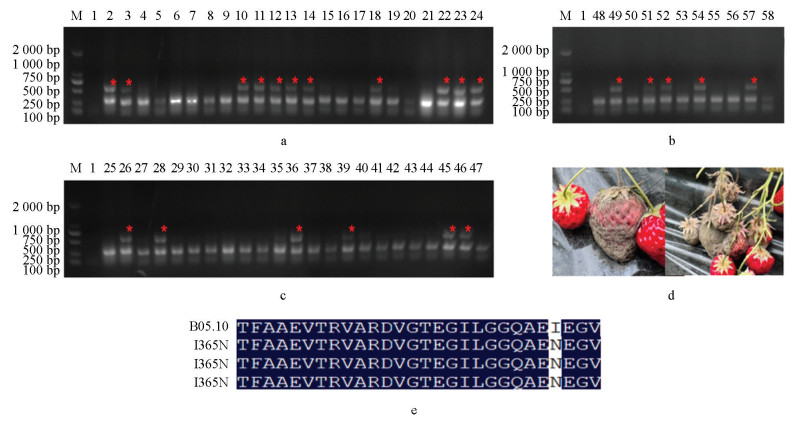
将田间采集的样本取样57个并粗提DNA为模板进行PCR检测,发现其中13个样本可在I365S检测体系中检测出2条PCR产物(图 7a-7c),抗性菌株比例为22.8%;22个样本可在I365N检测体系中检测出2条PCR产物(图 8a-8c),抗性菌株比例为38.6%,说明这些样本中可能分别含有I365S和I365N突变方式抗性菌株。然后扩增比对AS-PCR检测为阳性的部分菌株的BOS1基因,确证其抗性突方式分别为I365S(图 7e)和I365N(图 8e)。以上结果说明本AS-PCR检测体系可以快速准确地应用于田间灰葡萄孢菌二甲酰亚胺杀菌剂抗性。
2.1. AS-PCR特异性引物
2.2. 最适的退火温度
2.3. 多重AS-PCR内参引物浓度
2.4. 多重AS-PCR的特异性
2.5. 多重AS-PCR的灵敏度
2.6. 验证AS-PCR检测体系
2.7. 田间应用AS-PCR检测体系
-
由于缺少优良的抗病品种,目前灰葡萄孢菌主要以化学防治为主。二甲酰亚胺类杀菌剂(Dicarboximide Fungicides,DCFs)于20世纪70年代引入中国,随着该药剂的广泛使用,近些年在中国江苏[12]、四川[11]、辽宁[40]等地区都检测到了高频的DCFs抗性菌株。抗性检测技术的建立是防治的基础,为能够及时了解地区抗性发展情况调整用药策略,建立抗性检测技术是非常必要的。
传统抗性检测方法用时较长、工作量大、试验条件严格。随着分子生物学技术的发展,许多现代分子技术在抗药性检测中得到了很好的应用,特别是基于PCR的检测技术,如实时荧光定量、高分辨率溶解曲线法、双杂交探针法和限制性片段长度多态性等,但这些方法应用仪器精密度高、价格昂贵且步骤繁琐,并不适用于田间应用。等位基因特异性PCR技术具有操作简单、过程开盖次数少而污染概率低、特异性强、成本相对低(只需要一台PCR仪和一对特异性引物即可实现检测)、时间相对快速等优点,已经有许多研究者利用该技术成功检测出抗药性菌株,如Muñoz等[41]、Yin等[42]和Oshima等[22]分别成功利用该技术检测了灰葡萄孢菌苯对苯并咪唑类、琥珀酸脱氢酶抑制剂类和甲氧基丙烯酸酯类杀菌剂抗性菌株。但国内外尚没有应用该技术检测灰葡萄孢菌对DCFs抗性的报道。
前期研究发现,重庆地区灰葡萄孢菌DCFs抗性菌株中BOS1I365N(52.1%)和BOS1I365S(37.5%)点突变占比较大[28],因此,本研究建立的多重AS-PCR检测体系对于监测灰葡萄孢菌对DFCs抗性频率具有较好的应用价值。
本研究成功建立了可快速检测灰葡萄孢菌对DCFs抗性的AS-PCR技术,最终确定为3组引物I365S-7F/I365-R、I365S-8F/I365-R和I365N-8F/I365-R可分别特异性区分BOS1I365S和BOS1I365N点突变菌株,并对反应条件和反应体系进行优化,提高了该检测技术的特异性和扩增效率,最后应用于田间样本快速检测,证明该方法可行。
在引物设计时,需在突变点前后1~2个碱基进行错配[43],是因为DNA聚合酶理想情况下需要在引物与模板完全匹配的情况下才能顺利扩增,但实际情况中引物3'末端与模板不互补也能实现延伸扩增。错配不同会影响最终扩增效率和特异性,如罗梅等[36]在进行引物筛选时,引入G-A错配时无法区分敏感和抗性菌株,但G-C错配可实现区分且扩增较稳定,当错配2个碱基时扩增效率低。本研究中G-C错配可很好地实现区分,且错配为2个碱基AA-TG时也可明显区分且扩增效率尚可,说明建立检测体系可能因引物靶标序列和引物长度的不同具有差异性,所以在建立检测体系时需要根据具体靶标来设计错配并经过重复试验反复验证,从而筛选出能够特异性检测出抗性菌株的引物对。
为保证试验数据的可靠性,引入内参引物进行多重AS-PCR,可选择真菌通用引物[37]或其他非靶标基因[36, 44]。本研究选用前者,常用引物对为ITS1/ITS4,该内参引物在灰葡萄孢菌中扩增产物与特异性引物扩增产物相差小,电泳无法区分,通过ITS区内参引物筛选,最终确定ITS4/ITS86引物作为内参进行多重PCR。内参引物的引入可以很好地排除假阴性的干扰,且节约了试剂、DNA模板等原料。但由于不同引物在同一体系中同时扩增存在模板和试剂竞争,所以在多重PCR扩增过程中需要调节特异性引物和内参引物浓度比例。本研究发现特异性引物和内参引物的比例为4∶1时条带较浅,且由于人工操作重复加入的每个相同体系时存在的误差会导致该引物扩增不出产物,所以最终选择特异性引物和内参引物ITS4/ITS86浓度比例为2∶1。
特异性检测表明,该检测技术可以准确地从同种或不同种群体中检测到DCFs抗性菌株,具有很高的特异性。为能够更贴近田间实际应用,将CTAB大量精提DNA的方法替换为少量粗提的NaOH裂解法,将田间菌株在不进行分离纯化情况下粗提DNA作为模板进行PCR检测,扩增出特异性条带的菌株为BOS1I365S、BOS1I365N点突变,且该检测结果与测序结果一致。这简化了病原菌DNA提取步骤,缩短了操作时间,使得检测更加简单快速。
Muñoz等[41]设计2对引物并成功检测灰葡萄孢菌抗苯并咪唑类杀菌剂β-微管蛋白E198A点突变菌株。每对引物其中1条为3'末端位于突变位点上的内引物,两条引物方向相反,且分属于不同基因型(突变序列与野生型);另外两条为通用外引物,且两条引物与突变位点的距离不一样。这样,在反应过程中,无论检测模板为突变型或者野生型,其扩增产物都为两种,但产物片段长度不一,因此,根据扩增出的片段长度即可辨别该菌株是否为突变型,但该体系只能检测1种突变方式。目前,本研究通过设计2对引物分别在2个体系内将BOS1I365S和BOS1I365N点突变菌株进行区分,可采取上述Muñoz等[41]的方式同时加入内参引物,即可在一个体系内排除假阴性干扰的情况下又能同时检测2种突变方式。