


 下载:
下载:
-
开放科学(资源服务)标识码(OSID):

-
栽桑养蚕是我国先民的伟大创举,有着超5 000年的历史。蚕桑产业作为我国特色优势产业,在脱贫攻坚进程中发挥了关键作用,是当前乡村振兴的优势特色产业之一。培育高产且优质的蚕品种是推动养蚕业健康发展以及增加蚕农收入的重要根基。基因编辑技术在农业育种方面的应用越发广泛,不仅为作物抗病性的改良给予了关键支撑,还极大地推动了作物品质的提升[1-2]。在家蚕育种领域,转基因技术也被广泛用于创制优质育种素材[3-7]。就转基因素材来讲,明确其插入位点对其育种用途和推广极为关键。此外,家蚕亦是重要的实验动物,基于家蚕展开的功能基因研究对理解昆虫发育[8]、遗传[9]以及进化[10]有着重要的参考价值。转基因技术同样是功能基因研究的重要方式,确定转基因材料中目的片段的插入位点对明确目的基因的功能是不可或缺的。所以,构建高效、精准的鉴定家蚕转基因位点的方法对家蚕育种和功能基因的研究有着重大意义。
传统上,常用DNA印迹杂交[11]确定转基因材料中目的片段的拷贝数,再通过以聚合酶链式反应(PCR)为基础的方法确定插入位点,后者包括反向PCR、交错式热不对称PCR、质粒拯救法、外源接头PCR以及Sanger测序等。例如文献[12]利用反向PCR方法成功找到Bt11转基因玉米的侧翼序列;文献[13]利用交错式热不对称PCR方法成功分离了山药Pal和Pgi基因的5'-侧翼区域;文献[14]利用质粒拯救法成功在水稻中获取到插入T-DNA序列。然而,这些基于PCR的鉴定方法往往过程繁琐。例如,反向PCR方法需要将转基因系基因组酶切处理并环化,再经数轮巢式PCR扩增,才可能鉴定到插入位点[15];交错式热不对称PCR方法需采用温度梯度和不对称的引物设计,以增加扩增特异性[16];质粒拯救法需使用限制酶对DNA进行处理,并将其导入大肠杆菌内,使用PCR筛选阳性菌落并测序[17]。其他方法也涉及较多关键中间环节,使得传统基于PCR的鉴定方法不仅检测效率低、对实验人员技术要求较高,且其成功率不高,部分情况下需重复多次才能完成插入位点鉴定。综合来看,传统转基因位点鉴定方法操作复杂、特异性低,且受连接效率、退火温度、引物设计因素等限制[18]。随着以转基因技术为代表的分子育种技术的广泛应用,有必要在各物种中建立更高效、更精准的转基因位点鉴定方法。
近年来,随着高通量测序技术的普及,个体基因组测序成本迅速降低,使得利用基因组测序技术高效鉴定转基因插入位点成为可能。目前,在拟南芥[19-20]、玉米[21-22]、大豆[23-24]、水稻[25-26]、小鼠[27-28]、猪[29-30]、花椒树[31]、月季[32-33]等的研究中皆有利用基因组测序技术进行转基因位点鉴定的报道,但在家蚕的研究中尚未有过相关研究和报道。
本研究利用全基因组测序技术,对两个转基因家蚕品系的插入位点进行鉴定,建立一套快速确定家蚕转基因插入位点的方法。此外,还分析了测序深度如何影响插入位点检测的准确性。
全文HTML
-
转基因材料TransGene_1和TransGene_2是本实验室前期构建的2个过表达家蚕品系。TransGene_1:利用同源重组的办法,将目的基因表达核连接到piggyBac-{3×P3-EGFP}载体上,通过胚胎显微注射技术注射至新产非滞育蚕卵(G0代);随后,饲养G0代至化蛾(G1代),并在G1代蚁蚕刚孵化时筛选复眼发出绿光的个体;再饲养自交产生G2代,逐代筛选,最终获得纯合的转基因过表达品系。TransGene_2:将目标基因与家蚕A4启动子、SV40终止子通过中间载体pSL1180构建表达核[A4-Gene-SV40],酶切回收表达核后与piggyBac-[3×P3-EGFP]载体连接;再通过显微注射技术将重组载体转入家蚕非滞育卵(低温催青卵);最后利用荧光显微镜筛选复眼有绿色荧光的G1代个体逐代自交饲育,逐代筛选,最终得到转基因过表达品系。
使用TransGene_1和TransGene_2的蛹提取基因组DNA,使用血液/细胞/组织基因组DNA提取试剂盒建立DNA文库(百迈客生物科技有限公司),利用HiSeq X平台进行双端测序,每条reads长150 bp。
-
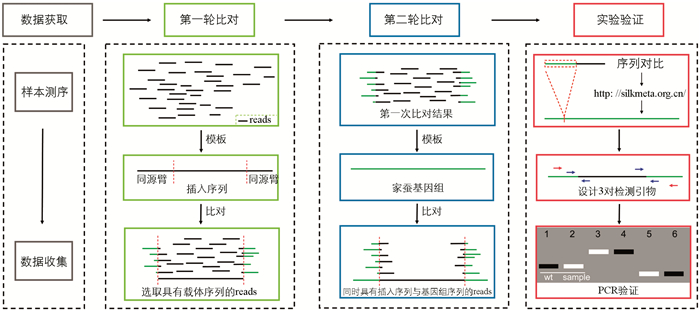
为了获得高质量的基因组测序数据用于后续研究,采取了以下步骤:①剔除N碱基含量超过5%的reads;②剔除质量值低于20且比例超过30%的reads。第一轮比对选用插入序列为参考,首先利用软件BWA(Burrows-Wheeler-Alignment Tool)v0.7.17将clean reads比对至参考序列;随后,使用软件samtools v1.6[34]对结果进行排序与筛选。第二轮比对选用家蚕基因组Bomo_genome[35]为参考基因组,以第一轮比对结果为输入进行比对,同样对结果进行排序与筛选。使用结果中的部分reads提交至网站
http://silkmeta.org.cn/ [36]进行BLAST分析,以获得具体的插入位置。 -
根据reads对比结果确定转基因载体的插入方向,拼接转基因材料序列,并设计引物加以确认(表 1)。引物设计原则如下:设计3对引物,其中2对为断点检测引物(即一条引物设计于基因组上,另一条设计于载体(左臂或右臂)),剩下一对为基因组检测引物,上下游均位于基因组上。借助凝胶电泳情况以及PCR产物测序结果,验证插入位点鉴定的准确性。
-
利用软件seqtk对双端测序文件予以随机抽取,从而模拟不同深度的测序状况,随机种子随机生成,每个深度设置11个重复。本研究总共设置9个梯度,模拟测序深度从2×至16×的情形。
1.1. 样本与基因组测序
1.2. 数据处理
1.3. 准确性验证
1.4. 测序深度依赖性检测
-
本研究运用全基因组测序技术对家蚕转基因的插入位点予以鉴定,具体分为以下4个步骤(图 1):其一,针对测序结果开展质量控制与过滤操作,去除低质量以及不适于分析的测序结果,留存clean reads以作后续分析之用。其二,于第一轮比对期间,挑选带有载体序列的reads,将插入序列当作参考,运用clean reads与之进行比对并对结果加以筛选。经筛选后的结果可分为两类,一类是完全比对成功的reads;另一类是仅有部分序列能够比对上的reads,此类reads又可细分为比对上左臂和比对上右臂两种情况,这两种情况中的reads均为需要保留的reads。其三,在第二轮比对时,筛选出与参考基因组存在部分相同序列的reads。其四,在实验验证步骤里,利用与参考基因组比对上的最长部分进行序列对比操作,确定插入位点,并设计引物开展实验验证。
分析表明,TransGene_1的基因组测序总共获取了约9.76 GB的数据,其中包含32 639 026条clean reads,Q30达到98.02%。TransGene_2的基因组测序共得到了约11.18 GB的数据,涵盖37 394 695条clean reads,Q30为97.95%。这两份材料的测序深度均约为20×(表 2),测序质量均符合后续分析要求。
-
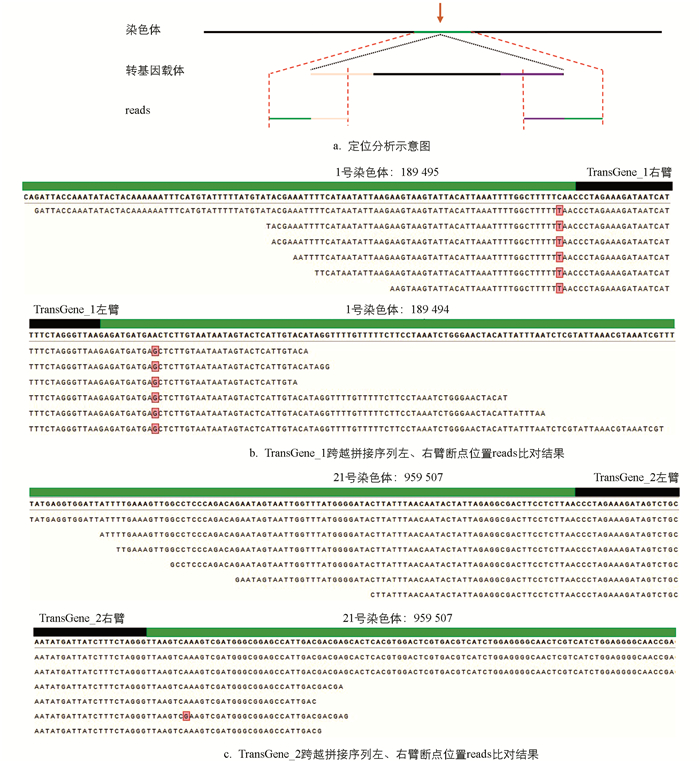
依据本研究的插入位点定位策略,初步判定转基因品系TransGene_1的插入位点处于1号染色体的1 899 494 bp之处,其插入方向为从同源右臂至同源左臂(图 2b);而转基因品系TransGene_2的插入位点位于21号染色体的959 510 bp之处,插入方向为从同源左臂至同源右臂(图 2c)。
为深入分析,对转基因材料的序列予以拼接和比对,且统计了可与家蚕参考基因组比对的reads的长度。在转基因品系TransGene_1中,最短碱基数为34 bp,最长碱基数为105 bp,平均碱基数为55.4 bp。而在TransGene_2中,最短碱基数为32 bp,最长碱基数为118 bp,平均碱基数为65.7 bp。获取具有可比对到家蚕参考基因组上的长碱基数的测序后读段(reads),有助于保证此分析方法比对结果的准确性(表 3)。
-
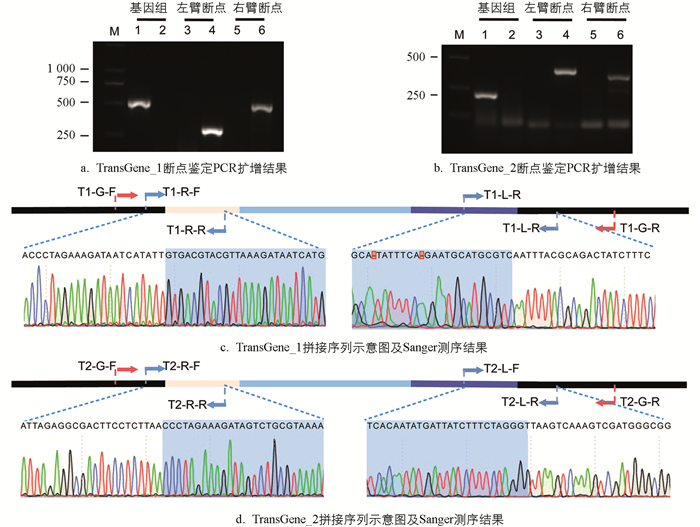
为了验证本实验方法的准确性,于转基因品系TransGene_1中设计了3对引物加以验证:左臂断点的扩增产物长度为304 bp,右臂断点的扩增产物长度为537 bp,野生型家蚕预计仅扩增590 bp的基因组序列。电泳结果表明:TransGene_1左、右臂断点产物的扩增长度和预期相符,并且未扩增基因组序列,这显示该品系为转基因纯合型;野生型家蚕仅扩增基因组序列,未扩增左、右臂断点,与预期一致(图 3a)。
在转基因品系TransGene_2中,同样设计了3对引物来进行验证,结果与预期相符(图 3b)。随后,对以上两个转基因品系的左、右臂断点扩增产物开展Sanger测序(图 3c、图 3d),结果和之前拼接的序列一致,验证了本研究方案的准确性。
-
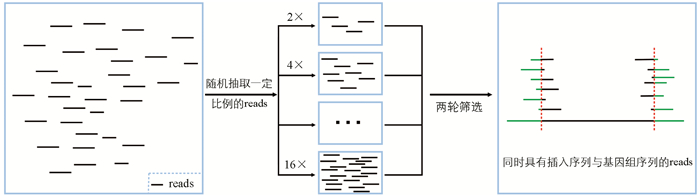
为深入探究测序深度对插入位点鉴定准确性的影响,本研究按一定比例对clean reads进行随机抽样,梯度设为2×、4×、6×直至16×,对抽取的reads开展两轮比对,每个梯度重复11次,以此模拟并比较不同测序深度下转基因插入位点的鉴定效率(图 4)。
利用两份转基因材料(TransGene_1与TransGene_2)的全基因组测序数据,对不同测序深度下转基因材料插入位点的鉴定情况进行了模拟(表 4)。结果表明,测序深度对插入位点的鉴定效果存在显著影响。测序深度为2×时,两份材料的未检测率分别为40.4%与18.2%,检测率分别为36.4%和18.2%,这表明此时鉴定效率较低。测序深度为5×时,两种载体的未检测率均降为0%,检测率分别提升至90.9%和72.7%,这意味着在此测序深度下鉴定效率有较大提高。测序深度为6×时,TransGene_1的鉴定率达到100%;测序深度为10×时,TransGene_2的鉴定率也达到100%,显示在这两个深度下鉴定效率分别达到最优。所以,本研究认为,针对两份转基因材料插入位点的鉴定,测序深度处于6×到10×之间,即可确保鉴定的有效性。
2.1. 插入位点鉴定策略与测序结果
2.2. 插入位点定位及序列比对分析
2.3. 插入位点鉴定准确性验证
2.4. 插入位点鉴定的深度依赖性分析
-
转基因生物的安全性一直是备受关注的焦点,涉及转基因操作技术安全问题与转基因产品安全性评估。在这方面,文献[37]已对转基因水稻可能给土壤质量和土壤生物带来的影响进行了深入研究。准确掌握转基因插入位点,对评估转基因生物安全性极为关键。本研究运用全基因组测序技术,针对两个前期构建的家蚕转基因品系开展了插入位点定位及后续验证工作。序列分析结果表明:TransGene_1的插入位点位于1号染色体1 899 494 bp与1 899 495 bp之间;TransGene_2的插入位点位于21号染色体959 510 bp与959 515 bp之间,这两个插入位点均属于基因间区。具体来说,TransGene_1插入位点距离上游基因KWMTBOMO00059 1 173 bp,距离下游基因KWMTBOMO00060 3 194 bp;TransGene_2插入位点距离上游基因KWMTBOMO12353 25 903 bp,距离下游基因KWMTBOMO12354 30 067 bp。
本研究借助全基因组测序技术精确鉴定了两个转基因品系的插入位点,并设计了检测引物,用于对转基因品系插入位点进行准确分型。这些检测引物不但可用于转基因品系的追踪与鉴别,而且可作为分子标记在其育种、选育中发挥作用。
-
本研究在测序深度为20×时,成功借助全基因组测序数据对插入位点予以鉴定,这表明在此测序深度下能够获取同时包含载体与基因组序列的reads。通常而言,较高的测序深度可提升捕获特定reads的概率[38],但相应地会使测序费用与分析成本增加。本研究探究了测序深度对位点鉴定准确性的影响。利用现有数据开展不同深度的模拟测试,结果显示,最低测序深度为6×时即可实现对插入位点的准确鉴定。有趣的是,本研究采用两份数据进行相同的模拟测试,然而另一份具有较多clean bases的材料却需在测序深度为10×的条件下方能完成鉴定任务,这或许是不同测序文库及插入位点的保守性存在差异所致。
在将全基因组测序与生物信息学手段相结合进行分析时,充足的高质量数据至关重要。文献[39]利用测序深度达182×的全基因组测序数据,针对快速循环育种中的转基因苹果T1109开展转基因插入验证,并与测序深度为167×的野生型苹果进行比较,结果表明在转基因苹果T1109中未检测到意外的转基因插入事件;文献[40]于测序深度为8×的转基因小鼠中成功鉴定出Pmel-1 TCR α和β转基因的基因组插入位点,这与本研究得出的最低测序深度(6×)相近。更高的测序深度能够获取更多的高质量数据,在运用生物信息学手段进行分析时能得到更精确的结果,但同时也可能致使假阳性结果增多。文献[41]对G2-6转基因水稻进行了T-DNA插入位点鉴定,在测序深度为70×时发现3条非水稻同源序列的reads,并通过实验证实这是由污染导致的假阳性。所以,在运用全基因组测序技术进行转基因位点鉴定时,应当结合物种特性以及插入基因的特点,选取适宜的测序深度。