


 下载:
下载:
-
开放科学(资源服务)标识码(OSID):

-
土壤中蕴藏着丰富的微生物,包括细菌、真菌、放线菌、古细菌、原生动物和线虫等,这些微生物群落是土壤生态系统的重要组成部分,对土壤生态功能和植物生长发育具有重要影响[1]. 大多数土壤微生物无法通过人工培养,传统的分离鉴定方法不仅费时费力,还只能检测可培养微生物. 宏基因组检测技术克服了传统方法的局限性,能够直接提供全面的信息. 近年来,随着宏基因组检测技术的应用,土壤微生物群落研究取得了显著进展. 宏基因组测序无需培养微生物,可帮助研究者全面了解土壤微生物的遗传信息和功能潜力,并能深入探究不同微生物的相互关系及群落的生态功能[2]. 宏基因组测序是一种用于检测土壤微生物群落的先进技术,能够提供全面的信息,包括遗传信息、多样性和功能潜力,并可深入揭示其结构和生态功能[3]. 它为土壤生态系统研究提供了全面而高效的方法,并为可持续土壤健康管理和生态保护提供了有力支持. 本文综述了近年来宏基因组学在土壤微生物群落研究中的应用技术和成果,以期为深入研究土壤微生物群落提供理论基础.
全文HTML
-
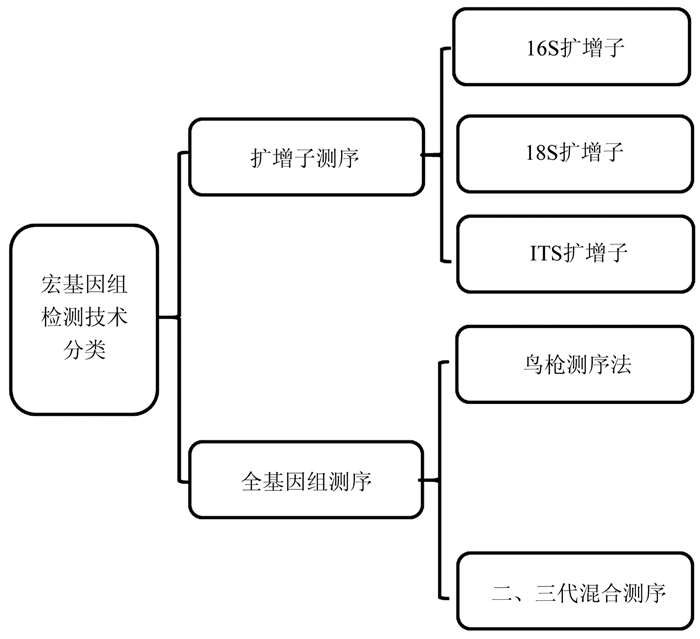
宏基因组检测技术主要研究环境中所有微生物群体遗传信息,可直接从环境样品中提取DNA,无需分离培养微生物,并利用高通量测序技术对混合DNA进行大规模测序,操作步骤包括采集环境样品、提取环境DNA、构建基因文库、高通量测序和数据分析等[4-9]. 在土壤微生物研究中,宏基因组检测技术展现了显著优势[10],与传统微生物学研究侧重于单一细菌或真菌的特定基因组不同,宏基因组学关注的是整个微生物群体的遗传信息,不仅可揭示已知微生物的存在,还能发现未知微生物的潜在新基因. 宏基因组学深入揭示了微生物群体的功能潜力,包括有益微生物的生态角色及其在土壤生态系统中的相互作用. 此外,宏基因组学还用于研究土壤微生物的适应性和响应机制,为理解土壤健康、生态系统功能及环境变化的影响提供了关键支持[11].
-
土壤微生物群落的物种多样性是指土壤中微生物物种的丰富度和相对丰度. α-多样性和β-多样性分析法是微生物群落研究中常用的分析方法,分别用于揭示群落内部多样性和不同样本间的差异. α-多样性描述单个样本内的群落多样性水平,常用指标包括Shannon指数、Simpson指数和Chao1指数,通过16S rRNA(细菌)或ITS(真菌)序列测序计算多样性指数. β-多样性主要比较不同样本间的群落组成差异,常用方法包括聚类分析和主坐标分析. 差异分析用于比较不同样本间微生物群落的差异,以揭示不同环境条件或处理方式对群落的影响. 常用统计方法包括ANOVA,Adonis,PERMANOVA等,可用于确定环境因素、生物样本来源等对群落组成的影响.
-
宏基因组学在基因功能预测中具有重要作用[12]. 宏基因组测序得到的DNA序列可与已知数据库(如NR、KEGG等)中的序列比对,通过比对蛋白编码基因的氨基酸序列,可以推断新序列的可能功能. 使用Clusters of Orthologous Groups (COG),euKaryotic Orthologous Groups(KOG)或Gene Ontology (GO)分类系统对宏基因组序列进行注释和功能分配,有助于预测新序列的功能,结合基因邻近关系、共表达和共现模式,可预测宏基因组序列参与的代谢通路和生物学功能.
2.1. 微生物群落物种多样性研究方法
2.2. 微生物功能基因研究方法
-
宏基因组学全面揭示了土壤微生物的物种组成,其多样性研究有助于深入了解土壤生态系统的健康状况、环境适应性及人类活动对微生物群落的影响.
-
相对丰度和多样性指数相互关联,共同揭示土壤微生物群落的结构和功能. Enagboma等[13]研究了白蚁丘土壤与周围土壤之间的细菌多样性差异,采用鸟枪测序方法对提取的宏基因组DNA进行测序,β-多样性分析显示白蚁丘与周围土壤样品之间的细菌功能类别存在显著差异. Bulgarelli等[14]采用16S rRNA基因测序与鸟枪宏基因组分析相结合的方法,分析了野生与驯化大麦相关的微生物群,结果显示丛毛单胞菌科、黄杆菌科和根瘤菌科在大麦根富集菌群中占据主导地位. 土壤中的微生物群落包括细菌、真菌和古菌等,其中细菌凭借高度的生存适应性和快速繁殖能力,在土壤中广泛分布且数量庞大. 细菌以异养型为主,少数为自养型. 真菌是土壤微生物群落中第二大类群. 古菌在土壤中的占比较低,但参与了硫循环等特殊的生物地球化学过程[15]. 总之,土壤中微生物的多样性及其相互作用对于维持土壤生态系统的平衡至关重要.
-
土壤微生物群落动态变化受到季节、植被类型、土壤性质和水分状况等多种因素的影响. 长期监测与宏基因组技术的应用使研究者能够追踪土壤微生物群落的时空变化. 分析不同土壤的宏基因组数据有助于准确揭示微生物群落的变化趋势,表 1为利用宏基因组技术检测土壤微生物群落变化的实例,并探讨了病害对群落多样性的影响.
-
土壤微生物多样性受自然和人为因素的共同影响[19-20]. 宏基因组学使用多元统计分析(如主成分分析、冗余分析)、机器学习(如回归模型、随机森林)、网络分析(如共现网络、功能关联网络)和功能预测(如基因功能注释、代谢途径分析)等方法来分析和预测环境因子(如温度、pH值、污染物)对微生物群落的影响[21-24]. 这些方法揭示了微生物群落结构和功能的变化趋势,为理解土壤微生物多样性的变化提供了支持. Tkacz等[25]采用高通量测序技术分析了4种植物的土壤、根际、根面和根内圈的原核和真菌特异性微生物群,结果显示,虽然细菌微生物群来自周围土壤,但其结构受根的影响大于土壤或植物物种的影响;相比之下,真菌微生物群主要受土壤的影响. Novello等[26]研究了位于意大利的一个综合虫害管理葡萄园中葡萄根际细菌微生物群的多样性,并比较了散装土壤和根际土壤的微生物群落结构,同时分析了开花和果实早期发育2个物候阶段对土壤微生物群落的影响;利用宏基因组学测序技术鉴定葡萄菌群后,结果表明生物多样性与物候期无关,但根际土壤的多样性高于散装土壤.
-
土壤微生物群落在有机物分解、养分循环、氮素转化及抑制病原体等方面发挥关键作用,并可提升植物免疫力. 同时,其作为生态系统的调节者,可促进植物健康、维持土壤生态平衡,并在农业生产和生态环境中发挥重要作用[27-31].
-
利用鸟枪宏基因组测序技术,Adedayo等[32]鉴定了健康蕃茄和白粉病番茄根际土壤微生物群落中与促生长和抗病功能相关的基因,结果显示健康根际微生物组中有21个植物生长促进基因,而病根际微生物组中仅含有9个,鉴定出的抗病基因包括核苷酸结合基因和抗菌基因.
-
土壤微生物群落的代谢潜力体现在降解有机物、促进养分循环和产生抗生素等功能上. 微生物通过这些代谢活动,在生态系统中扮演关键的功能角色. 利用宏基因组学检测技术研究土壤微生物群落代谢能力的实例见表 2.
-
研究土壤生态系统中的关键微生物种群(如氮循环微生物、磷溶解细菌和腐解真菌)有助于深入了解它们在养分循环和有机质降解等生态功能中的作用[36]. Jiao等[37]利用高通量扩增子和宏基因组测序发现,核心微生物群对于维持再造林生态系统中土壤微生物群的功能稳定性至关重要. 利用宏基因组测序技术研究关键微生物种群的实例见表 3.
3.1. 土壤微生物群落的物种多样性
3.1.1. 相对丰度和多样性指数
3.1.2. 土壤微生物群落的多样性变化
3.1.3. 影响土壤微生物多样性的因素
3.2. 土壤微生物群落的功能潜力
3.2.1. 功能基因的注释和预测
3.2.2. 土壤微生物群落的代谢潜力
3.2.3. 土壤生态系统功能的关键微生物种群
-
随着测序技术的不断发展,宏基因组学将提供更高分辨率的微生物群落解析,揭示更多细节和功能,并检测微生物群落的细微变化. 结合宏基因组学数据和计算模型,可构建微生物群落的动态模拟与预测模型,推测其在不同环境条件下的演变趋势. 宏基因组学检测技术可挖掘微生物群落中的未知物种和功能基因,推动新型生物农药、生物能源和生物材料等产品的开发. 通过整合宏基因组学数据与其他组学数据(如宏转录组学、宏代谢组学),可以更全面地理解微生物群落的功能和代谢活动,加深对微生物与宿主(包括植物)互作的认识. 随着技术创新和数据处理方法的提升,宏基因组学将成为解析土壤微生物群落结构和功能的重要工具,为可持续土壤管理和生态系统保护提供科学依据.