


 下载:
下载:
-
开放科学(资源服务)标志码(OSID):

-
伴随着页岩气勘探和开采技术的进步,日益丰富的天然气资源及其相对于其他轻质烷烃的潜在竞争优势,使得从天然气中生产化学中间体和高附加值的化学品成为一种有吸引力的选择[1-3]. 乙炔(C2H2)被誉为“有机合成之母”,它不仅在金属加工、焊接和切割领域发挥着重要作用[4-5],而且还用于生产其他化学品,如氯乙烯、乙醛、醋酸乙烯、丙烯腈和丙烯酸等产品[6-9],其广泛的应用和丰富的下游产品促使学术界和产业界对其生产方法更为关注.
Khan等人[10]对甲烷(CH4)热解的动力学参数进行了总结;Dean[11]提出了由25个物种和44个反应组成的简化模型来解释CH4的热解过程;Steinberg[12]研究了CH4在973~1 173 K条件下的热解动力学;Baranov等人[13]研究了CH4和氢原子混合物的热解,指出C2H2的快速形成是通过激发乙烷(C2H6)和乙烯(C2H4)分子的二次解离所引起的;Rodat等人[14]模拟了CH4在1 500~2 300 K下的热裂解,预测了气体浓度随停留时间的变化关系;Lümmen[15]利用反应力场分子动力学模拟研究了CH4热解过程;Paxman等人[16]研究了CH4热分解的动力学参数;Xue等人[17]利用反应力场分子动力学研究了CH4在不同温度和密度下形成纳米腔的过程;Dinh等人[18]研究了电弧法CH4直接转化为C2H2的过程;Ogihara等人[19]研究CH4和乙烷(C2H6)混合物在973~1 073 K下的热解.
总的来说,CH4热解制备C2H2过程已经受到广泛的关注,但目前多数报道都是在1 200~2 000 K温度下进行的研究. 而在实际天然气制备C2H2工艺中,广泛使用的是将天然气一部分用于燃烧,为余下天然气热解提供能量,燃烧和裂解在乙炔炉中同时进行. 天然气在燃烧的过程中会瞬间释放出大量的热量,促使局部高温出现,而此时会发生CH4向C2H2转化. 因此,研究CH4在高温下热解形成C2H2的反应机理是很重要的.
在以前的报道中,有关CH4热解过程中的CH3,C2H4,C2H2反应途径的敏感性分析是很少见的,所分析的反应也是有限的. 同时,热解反应速度非常快,会产生大量的中间体和自由基,这些中间体和自由基与温度、压力等热力学条件有关. 如果仅仅是从实验获得的最终产物去分析各种基本步骤是一项极其困难、艰巨的任务. 因此,在原子水平上对CH4高温热解制备C2H2的反应过程进行理论研究,阐明反应机理以及C2H2的主要形成和消耗途径是对实验研究的必要而有益的补充.
随着计算机科技的快速发展,分子动力学模拟方法已经在各领域得到了广泛的使用[20-22]. 在现有的反应力场中,由Adrivan Duin和William A Goddard Ⅲ设计的ReaxFF反应力场是使用最广泛的反应力场之一[23-25]. 该方法基于键级和键距的关系,可以准确地描述键的断裂和形成,有助于对化学反应进行原子和分子水平上的研究. 目前,反应力场分子动力学已经广泛应用于研究烃类的热解过程[26-31].
本研究结合密度泛函理论和反应力场分子动力学模拟,分析了CH4在不同温度和压强下热解制备C2H2的过程. 第1部分研究了温度、密度对CH4热解制备C2H2的影响,并结合数学分析得到了最适的反应温度和压强;第2部分研究了高温下CH4到C2H2的转化机理,分析了C2H6,C2H4,C2H2的主要形成和消耗途径,并与文献对比,发现了一些新反应途径. 这些研究有助于更全面、更系统地认识天然气转化为C2H2的反应过程,对天然气制备C2H2的工艺优化具有参考价值.
全文HTML
-
根据反应涉及物种的焓、熵和热容,评估纯CH4及其裂解产物在不同温度下的平衡组成. 采用密度泛函理论在B3LYP/6-311+G(2df,2p)水平上计算了不同温度和压强下的反应吉布斯自由能. 其中,温度范围为1 273~3 773 K,间隔100 K. 压强范围为1~30 MPa,间隔为0.5 MPa. 最后,通过数学分析得出CH4裂解制备C2H2的最优条件. 所有密度泛函理论计算均使用Gaussian 09程序进行,数学分析使用Wolfram Mathematica 8.0软件包进行.
-
Materials Studio (MS)用于构建模拟的初始结构,模拟盒子是由200个CH4分子组成,大小为4.78 nm×4.78 nm×4.78 nm,密度为0.05 g/cm3. 首先,温度在500 ps内均匀升到3 500 K,然后在3 500 K下保持500 ps的恒温,以研究CH4到C2H2详细的反应机理. 使用等温等压系综,模拟温度由Nosé-Hoover温控器控制,阻尼常数为100 fs. 模拟时间步长设置为0.1 fs. 所有分子动力学模拟使用Lammps[32-33]软件完成.
1.1. 量子化学计算
1.2. 分子动力学模拟
-
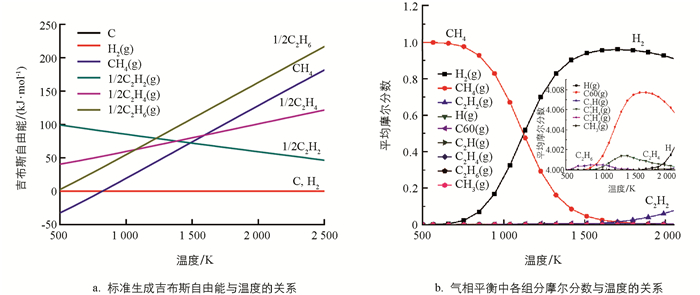
通过HSC软件研究CH4热裂解过程中涉及的主要物种的吉布斯自由能与温度关系以及平衡摩尔分数与温度关系. 如图 1a所示,碳和氢气的标准生成自由能为0. 在较高温度下,如果反应时间足够长,烃类裂解的最终产物只会是碳和氢气. CH4在低温(≤ 800 K)下是非常稳定,随着温度升高,CH4变得越来越不稳定. 当温度大于1 500 K时,CH4甚至比C2H2更不稳定,这表明温度在1 500 K以上,CH4裂解生成C2H2在热力学上是有利的. 同时可以看出,在高温时C2H2相对于碳和氢气仍然是不稳定的,意味着C2H2有进一步裂解为碳和氢气的趋势. 为了防止C2H2分解成碳和氢气,必须在一定时间后终止反应,这与已有实验研究结果[34]是一致的,实际工业生产中也采取淬冷的方法来终止反应.
运用最小吉布斯自由能函数法来确定平衡系统的组成. 以CH4为底物,温度范围为373~2 273 K,结果见图 1b. 热解过程中产生的主要物种是H2和C2烃类. 最先出现的烃是C2H6,在600~1 000 K左右有较多的累积,1 000 K后C2H6量开始减少;在900 K左右,随着烷烃C2H6的减少,烯烃C2H4在增加,大约在1300 K达到最大量的累积;这个计算结果与实验结果相一致[35]. 温度进一步升高时C2H4含量开始衰减,并开始产生C2H2,这与先前的结果是一致的[36].
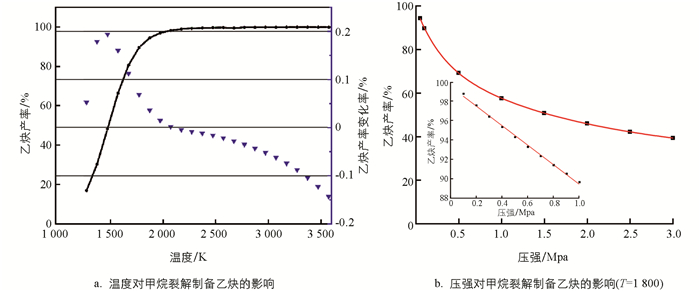
为初步评估CH4转化为C2H2过程中温度和压强对CH4转化率和C2H2产率的影响,本研究采用简化模型2CH4=C2H2+3H2进行分析. 如图 2a所示,随着温度升高,C2H2产率升高,但在1 500 K后,反应温度升高对C2H2平衡产率的提升效应开始减弱,综合考虑产率等因素,优选的温度区间应该是1 700~1 800 K. 如图 2b所示,对于压强,当小于大气压时,C2H2产率与压强呈线性关系;但随着压强的继续增加,C2H2产率整体上呈指数衰减趋势. 考虑反应速率、C2H2产率等因素,压强应控制在70~80 kPa之间.
-
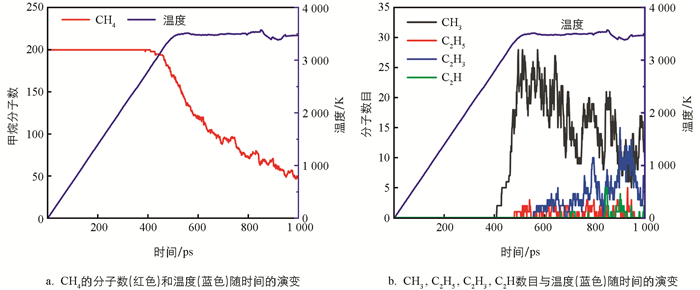
从图 3a中可以看出,CH4在2 800 K时开始裂解. 模拟结果表明,首先是发生反应CH4→CH3+H,CH4+H→CH3+H2. 即CH4的衰减主要是产生了CH3. 根据CH4的裂解速率,可将CH4的裂解分为3个阶段:第1阶段为410~460 ps(2 800~3 200 K),CH4的裂解速度相对较慢,只有4%的CH4分子被消耗;第2阶段为460~700 ps(3 200~3 500K),CH4的消耗速率增加;而第3个阶段在700 ps以后,CH4分子数呈波浪式下降.
图 3b展示了CH3,C2H5,C2H3,C2H数目和温度随时间的变化. 可以发现,随着CH4的裂解,CH3的量并没有持续地累积,而是处于在某一个值并上下波动,表明CH3要继续参与反应. 而体系中的C2H5自由基是较早出现的,但其积累量非常低. C2H3自由基在C2H5自由基之后产生,在C2H自由基之前产生,并且在反应过程中有明显的积累量,出现了一个较大的峰值. 在整个过程中,C2H最后出现且量是很少的.
-
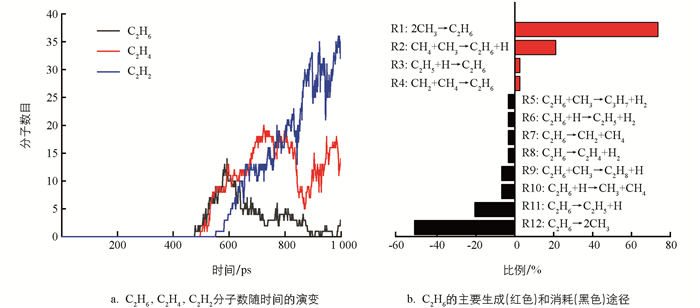
在热解过程中有烷烃、烯烃、炔烃、氢气和大量不稳定的自由基. 总的来说,主要是含C2的烃类和氢气. 因此,接下来主要是对C2烃类物种相关反应进行分析和讨论. 通过图 4a发现,C2H6的量在500~650 ps内变化明显,后期处于一个值并上下波动. 因此,重点分析这期间内C2H6的变化,时间间隔为1 ps. 500 ps后生成C2H4和C2H2. 为了简化分析,在下面的讨论中,以500~1 000 ps,使用10 ps间隔的统计数据来获得随时间变化的反应分布.
在CH4热解中,C2H6是最先形成的烃类,且量比较丰富. 通过对其轨迹文件进行分析,C2H6的产生与消耗所涉及的反应见图 4b. 很明显,C2H6的主要产生途径源自CH3的二聚(R1:2CH3→C2H6),次要的形成途径源于CH4和CH3的碰撞(R2:CH4+CH3→C2H6+H). 在热解过程中,形成的C2H6继续参与反应,均裂产生两个CH3,脱氢将生成C2H5,这是C2H6的主要两个消耗途径. 对比可以发现,C2H6脱氢生成C2H5的比例远远高于C2H5吸氢形成C2H6的比例. 这暗示C2H6在反应过程中主要表现为脱氢.
将模拟中观察到涉及C2H6的反应与已有的反应机制进行对比,并在m062x-D3/def2TZVP水平下计算反应在298 K和3 500 K下的吉布斯自由能变ΔG,结果见表 1. 通过表 1可以发现,与文献对比后,在反应力场分子动力学模拟中还发现CH4与CH2碰撞将形成C2H6,C2H6与CH3碰撞会形成C3H8或C3H7. 在以前的报道中[15, 37],只强调了反应R2是产生C2H6最主要的途径,忽视了反应R1对C2H6形成的贡献. 同时,对于C2H6消耗的途径,只突出了C2H6自身脱氢反应的主导地位,忽视了其自身的均裂(R12). 这可能是温度所引起的,正如表 1所示,反应R1在低温下较容易进行,而反应R12在高温下ΔG为负值,反应较易发生. 因此在模拟中反应R12是C2H6主要消耗途径是合理的.
-
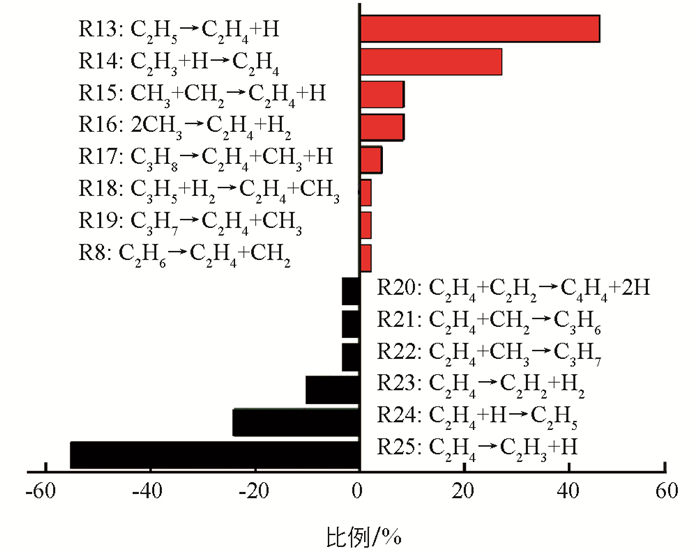
C2H4是CH4热解过程中最丰富的烯烃,出现于C2H6烷烃之后. C2H4的产生与消耗途径见图 5. 从图 5可以发现,C2H4的主要产生途径是C2H5裂解脱氢(R13:C2H5→C2H4+H),其次是C2H3的吸氢(R14:C2H3+H→C2H4). 这两个反应的逆反应(R24和R25)也是C2H4的两个主要消耗途径. 即C2H4脱氢产生C2H3比例比吸氢形成C2H5的比例高很多,整体上表现为脱氢. 这与体系中的C2H5数量一直处于一个比较低的结果相一致.
将涉及C2H4生成和消耗的反应途径与已有反应机制进行对比,结果见表 1. 除能观察到与文献一致的反应外,还发现了一些新的化学反应. 如:C3H8和C3H5裂解后将形成C2H4;C2H4与CH2碰撞后将形成环烷烃C3H6;C2H4与C2H2反应将生成高度不饱和烃C4H4. 以前的文献[15, 37]指出反应R13是形成C2H4的主导途径,反应R16也将促使其生成,以及反应R24和R25是C2H4的2个主要消耗途径. 但在模拟中发现,反应R14和R15对形成C2H4的贡献也是不可忽视的. 同时对C2H4的消耗反应R23的同样也是不可忽略的. ReaxFF模拟表明,C2H3的吸氢对形成C2H4是很重要的,C2H4的吸氢也将导致其数量上的衰减. 在实际生产中,C2H4也是一种重要的化工产品,阐明纯烃体系中C2H4的生成和消耗途径以及它们发生的比例将有助于优化C2H4工艺.
-
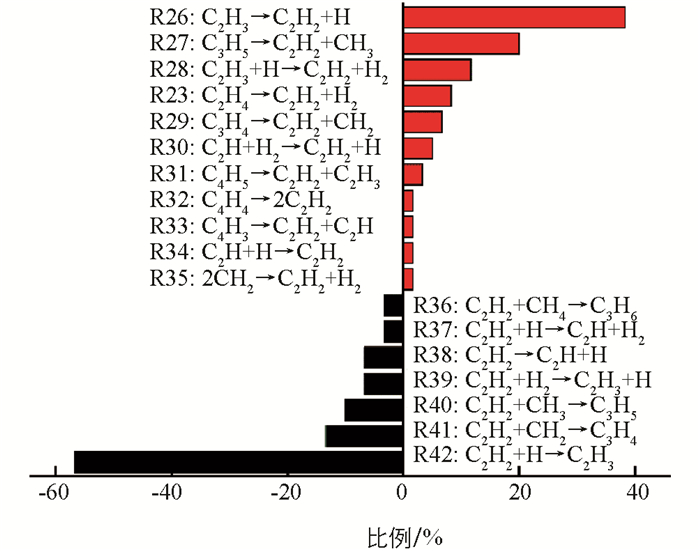
C2H2是模拟中丰度最大的炔烃,产生于烷烃和烯烃以后. 如图 6所示,C2H2的主要形成途径是C2H3的裂解脱氢(R26:C2H3→C2H2+H),其次是C3H5的裂解(R27:C3H5→C2H2+CH3),分别占38.3%和20.0%. 前者的逆反应(R42:C2H2+H→C2H3)也是C2H2的主要消耗途径. 即C2H2易与体系中的H自由基碰撞结合产生C2H3,而C2H4主要消耗途径也是自身脱氢产生C2H3. 可以发现,体系中C2H3形成途径不是单一的,因此C2H3含量会随时间出现较大的波动,该结果与图 3b是一致的. 对比发现,C2H2的形成途径较多,包含CH2之间的碰撞、C2Hn(n=3,4)的脱氢,C3Hn(n=4,5)中的C—C键断裂以及C4Hn(n=3,4,5)的裂解. 这可能也是CH4高温裂解中C2H2含量在某个阶段相对较多的原因.
在模拟过程中发现了一些新的化学反应途径. 如:C3H4和C4H4在发生C—C键断裂后将形成C2H2;C2H2与H反应也将生成C2H和H2;C2H2与CH4碰撞后将形成丙烯C3H6. 以前的报道[15, 37]指出反应R26是形成C2H2的主导途径,反应R23是其次要形成途径,对于C2H2主要消耗途径是反应R40. 但在模拟中发现,反应R27是形成C2H2的次要途径,同时也观察到反应R42和R41对C2H2消耗的贡献. 正如表 1所示,反应R27在常温下的ΔG为正值,在高温下为负值. 因此,反应R27在高温下更易进行,而逆反应R40在常温下更易进行. 即C2H3的脱氢和C3H5的裂解对形成C2H2是很重要的,C2H2的吸氢以及与CH2和CH3的反应将导致其数量上的减少.
-
在3 500 K时,反应初期主要是形成CH3. Marques等人[45]的研究显示,在3 500 K时,CH4热解为甲基和氢的速率常数k为2.5×1013/cm3(mol·s);在考虑零点能效应后,k为2.1×1010/cm3(mol·s),通过分子动力学获得的k值应该位于这两个数值之间. 本研究的模拟结果显示,3 500 K时,相应反应的速率常数为1.23×1013 cm3/(mol·s),与文献结果是一致的.
2.1. 量子化学计算结果与分析
2.2. 分子动力学模拟结果与分析
2.2.1. CH4的裂解反应
2.2.2. C2H6的相关反应
2.2.3. C2H4的相关反应
2.2.4. C2H2的相关反应
2.3. CH4热解速率常数
-
结合密度泛函理论和反应力场分子动力学模拟,对CH4高温下裂解制C2H2反应过程进行深入研究. 主要结论为:①高温、低压有利于CH4转化为C2H2;CH4制备C2H2的最优温度是1800 K,最优进料初始压强为70~80 kPa. ②温度为3 500 K时,CH4热解的速率常数为1.23×1013 cm3/(mol·s). ③ CH3的二聚和CH4和CH3的碰撞是形成C2H6的主要途径,C2H6均裂形成CH3和脱氢生成C2H5的反应是造成C2H6消耗的主要原因. ④对于热解产生的C2H4,其主要形成途径是C2H5裂解脱氢和C2H3的吸氢;其主要消耗在于自身裂解脱氢形成C2H3以及吸氢生成C2H5的反应. ⑤对于产物C2H2,其形成途径较多,占主导地位的是C2H3裂解脱氢和C3H5的裂解;C2H2主要消耗途径在于自身的吸氢以及与CH2和CH3的反应. 本研究通过ReaxFF分子动力学模拟CH4高温下热解制备C2H2的反应过程,还发现了一些新的反应通道. 通过进行敏感分析,找出对产物占主导地位的反应途径,这对天然气裂解制备C2H2的工艺优化具有参考价值,同时有助于建立更完善的CH4裂解反应机制.