




-
开放科学(资源服务)标志码(OSID):

-
阿尔茨海默病(Alzheimer's disease,AD)是最常见的一种神经退行性疾病,其临床特征为患者出现认知功能,特别是记忆功能的损伤,并伴有各种神经症状和行为障碍[1]. 目前,全球约有3 300~3 850万AD患者,其中我国患者超980万,因患者长期接受治疗及护理产生的巨额费用给社会造成沉重负担[2-3],因此,AD的诊断、预防和治疗一直是全球范围内关注度极高的研究领域.
AD作为一种成因复杂的系统性疾病,人类至今仍未能明晰其确切的病理成因,以中枢胆碱能假说为基础设计的胆碱酯酶抑制剂目前仍是AD临床治疗的首选策略之一. 该假说认为,AD患者脑内,特别是皮质和海马、前脑Meynert基底核和隔区等部位的胆碱能神经元受损严重,这导致以乙酰胆碱(acetylcholine,ACh)为代表的重要神经递质的水平与功能严重降低,进而导致认知功能障碍等一系列典型的AD病理特征. 胆碱酯酶负责完成ACh的水解与代谢,是控制其含量水平的关键因素,包括乙酰胆碱酯酶(acetylcholinesterase,AChE)和丁酰胆碱酯酶(butyrylcholinesterase,BuChE)[4]. 因此,胆碱酯酶抑制剂可通过抑制AChE或/与BuChE的活性,增加突触间隙处ACh浓度,增强中枢胆碱能神经传递能力,提高ACh的生物利用度,从而改善患者的认知与行为症状. 目前仅有4个胆碱酯酶抑制剂(他克林、多奈哌齐、加兰他敏、利凡斯的明)经美国食品药品监督管理局(FDA)批准进入临床,其中他克林由于存在肝毒性已撤市,因此急需研究新型的胆碱酯酶抑制剂.
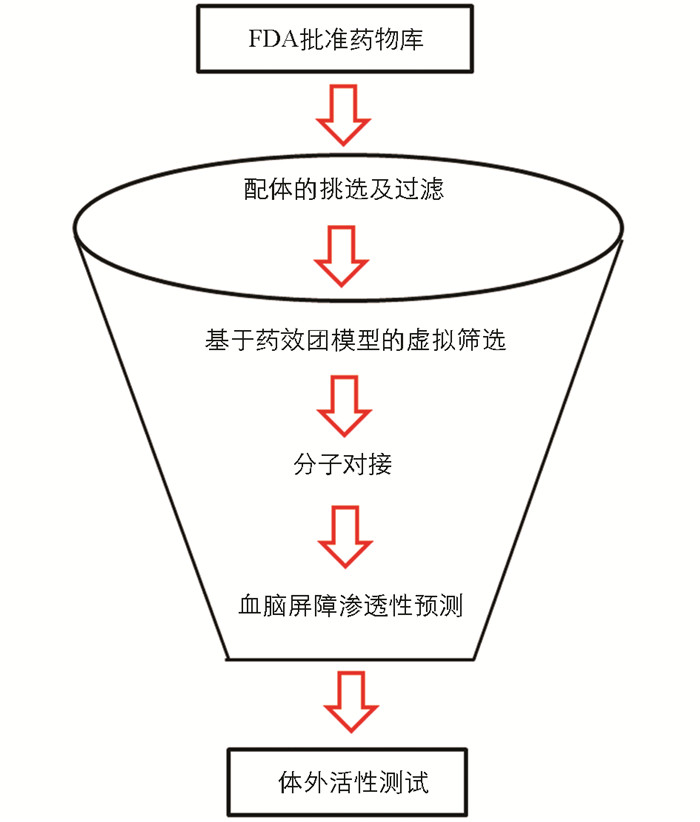
近年来,随着多个AD候选药物在临床阶段折戟沉沙,巨大的投入与产出间的失衡使得药物重定位策略越来越受到科学界与工业界的重视. 药物重定位,又称老药新用,指对已上市药物或者未上市但结构明确、生物活性已知的药品,通过进一步研究发现其新的靶点,扩大其适应症范围[5]. 与从头研发获得首创(first-in-class)新药相比,上市药物由于已经通过长期的临床实践,其毒性与不良反应均已明确,且生产工艺、质量标准、剂型等业已完备,因此在新药研发的时间和经济成本方面具有显著的优势. 本研究采用“基于结构的虚拟筛选”策略以及“老药新用”策略,综合运用多种计算机辅助药物设计技术对FDA批准药物库进行虚拟筛选,以挖掘新型胆碱酯酶抑制剂分子,为抗AD药物的研究提供新的思路和研究基础(图 1).
HTML
-
从ZINC数据库(https://zinc.docking.org/substances/subsets/fda/)下载经FDA批准的上市药物,从RCSB PDB数据库(https://www.rcsb.org/structure/5NN0)下载人源AChE与抑制剂的复合物晶体结构(PDB ID:4EY7)以及人源BuChE与抑制剂的复合物晶体结构(PDB ID:5NN0)[6-7]. 分子模拟软件采用Schrödinger 2019-1(Schrödinger,LLC,New York,USA).
-
使用Schrödinger软件中的Protein preparation wizard对下载的复合物晶体结构进行准备,包括加氢、加电荷、优化loop区、进行能量最小化等[8]. 使用LigPrep对下载的FDA批准上市药物分子(配体库)进行准备以获得高质量的3D结构,包括加氢、加电荷、生成同分异构体、手性异构体等[9]. 使用Ligand filtering对3D结构进行过滤,参数设置为Charged_amines≥1.
-
药效团的构建使用Schrödinger软件Phase模块中的Develop pharmacophore model[10-11]. 参数设置:Create pharmacophore modeling using选择Receptor-ligand complex,Method选择Manual,Choose ligand选择92H(627),Hypothesis settings选择Create receptor-based excluded volume shell. 获得初步药效团模型后,对药效特征元素进行编辑,Usage in screening选择Required.
基于药效团的虚拟筛选采用Phase模块中的Phase ligand screeing进行[10-11]. 在Screening settings中对筛选过程中产生的构象数目进行定义:Generate conformers during search为200.
-
采用Schrödinger软件中的Glide模块进行分子对接[9],采用模块中的Receptor grid generation对蛋白的对接位点进行定义,参数设置:Define receptor选择Pick to identify the ligand,Size选择Dock ligands with length≤20Å. 采用模块中的Ligand docking进行级联分子对接,参数设置:Precision先后选择SP(standard precision)和XP(extra precision),每个配体报告的最终构象数目定义为Write out at most 10 poses per ligand.
-
采用Schrödinger软件中的QikProp程序进行BBB渗透性预测,所有参数采用默认设置.
-
从Topscience(www.tsbiochem.com)获得3个药物(米托蒽醌,帕比司他,赖诺普利),纯度大于95%(LC-MS). 采用优化后的Ellman法测定化合物的胆碱酯酶抑制活性. AChE(来自人红细胞,C0663),BuChE(来自马血清,C7512),5,5'-二硫代双(2-硝基苯甲酸)(DTNB,D218200),碘代硫代乙酰胆碱(ATC,A5751)以及碘化丁酰硫代胆碱(BTC,B3253)购自Sigma-Aldrich(St. Louis,MO,USA). 测试过程中,先将10 μL AChE溶液(2.5 units/mL)或BuChE溶液(2.5 units/mL),20 μL DTNB溶液,10 μL测试化合物溶液加至40 μL缓冲溶液(pH值为8.0)中,加入20 μL ATC或BTC溶液触发反应后开始计时并快速混匀测试溶液,2 min后于412 nm波长下使用赛默飞酶标仪(Multiskan FC,51119080,USA)测量紫外吸收度. 空白对照组用10 μL水代替酶溶液. 通过公式计算抑制率(I)
式中,Ai,Ac和Ab分别代表测试组、阴性组与空白组对应的吸光值. 配制测试化合物的浓度梯度溶液(10-5~10-10 mol/L),记录受试化合物在各个浓度的吸光值,所得结果用GraphPad Prism 6(GraphPad Software,San Diego,CA,USA)计算IC50值. 所有测试重复3次,数据以x±s表示.
1.1. 数据及模拟软件
1.2. 蛋白及配体库的准备
1.3. 药效团的构建及虚拟筛选
1.4. 分子对接
1.5. 血脑屏障(BBB)渗透性预测
1.6. 体外胆碱酯酶抑制活性评价[12]
-
ZINC数据库作为公开数据库,收集了大量商业可得的化合物用于虚拟筛选研究[13]. 从该数据库中获得了1 615个经FDA批准上市的药物分子的3D结构,采用Schrödinger软件中的LigPrep对原始结构进行准备,包括加氢、加电荷、产生同分异构体等. 研究已表明,胆碱酯酶的活性位点峡谷具有相似的特点,均由外周阴离子位点、乙酰结合口袋、胆碱结合口袋以及催化活性位点组成. 带负电荷的外周离子位点在吸引正电荷底物和抑制剂进入活性位点峡谷,并到达距离入口处20Å的催化活性位点过程中扮演着重要作用[14-15]. 此外,结构生物学研究也表明,具有正电荷的配体能够同活性位点内的芳香氨基酸残基形成阳离子-π相互作用. 因此,多个已报道的活性AChE,BuChE抑制剂均具有含氮碱性基团以及形成正电荷中心的能力. 基于上述原因,本研究中在完成配体准备后,使用Ligand filtering对带电荷的胺进行挑选以提高命中率,共获得664个构象.
-
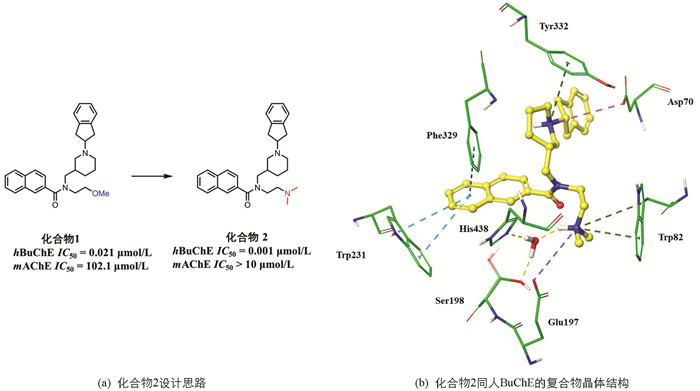
药效团是以药效特征元素为基础建立的模型,基于药效团的虚拟筛选在药物及先导化合物发现中扮演着重要角色. 近年来,多个人源胆碱酯酶蛋白与抑制剂的复合物晶体结构被报道,为开展基于结构的虚拟筛选研究提供了物质基础[16-20]. 2018年,KOŠAK等[6]在前期研究的基础上开展基于结构的合理药物设计(图 2a),获得了一个高活性高选择性BuChE抑制剂(化合物2)及其与蛋白的复合物晶体结构(PDB ID:5NN0,分辨率为2.1Å). 相较于其鼠源AChE(mAChE)抑制活性,化合物2对人源BuChE(hBuChE)的体外抑制活性达到了低微摩尔水平. 图 2b中,抑制剂的萘环能够同乙酰结合口袋中的Trp231以及Phe329产生T形π-π堆积作用. AChE中的乙酰结合口袋体积更小,因此该处具有较大空间位阻的萘环也许能够解释抑制剂具有高BuChE选择性的原因. 哌啶环上的氮形成了一个正电荷中心,同邻近Tyr332的苯环产生了阳离子-π相互作用. 同时,还与外周位点处Asp70形成了盐桥. 此外,抑制剂的二甲氨基乙基侧链伸向胆碱结合口袋,与胆碱结合口袋内的Trp82形成阳离子-π相互作用,并同Glu197形成盐桥. 值得注意的是,水分子(HOH805)通过桥联作用在抑制剂分子与组成催化位点的His438,Ser198间建立起了联系.
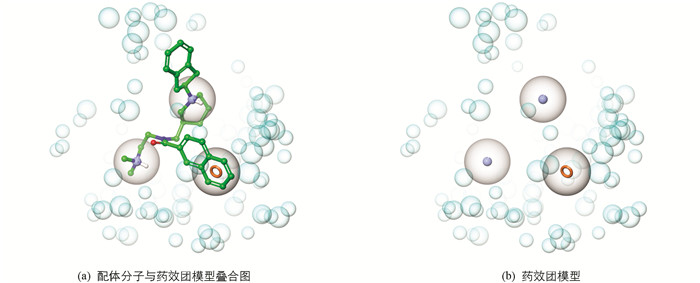
基于上述结合模式分析结果,采用Schrödinger软件Phase模块中的Develop pharmacophore model,手动构建用于虚拟筛选的药效团模型:①将萘环中的1个苯环定义为芳香环药效特征元素;②将哌啶环上的氮定义为正电荷药效特征元素;③将侧链最末端的二甲氨基定义为正电荷药效特征元素. 同时,基于受体结构中的原子产生排除体积,获得了初步的药效团模型(图 3),模型包含3个药效特征元素,蓝色实心球代表正电荷药效特征元素,橙色空心环代表芳香环药效特征元素,以及基于受体原子产生的排除体积(淡蓝色空心球). 考虑到药效团模型中药效特征元素的数量仅为3个,因此本研究对药效特征元素在筛选时的角色进行了约束,要求配体必须同时满足所有药效特征元素才能被保留. 对挑选获得的664个构象进行基于药效团的虚拟筛选,获得48个构象(Align score>0.2).
-
分子对接是结构分子生物学以及计算机辅助药物设计中的关键工具. 通过分子对接研究人员可以对配体-蛋白结合模式以及配体对蛋白的理论亲和力等进行预测. 在本研究中,采用Schrödinger软件中的Glide模块进行grid文件的准备以及分子对接研究. 通过分析结合模式以及调研文献,删除了除水分子724,734,805以外的其余水分子,随后通过重对接对grid文件以及对接软件(方法)进行考察. 重对接结果表明,当代表性对接构象(Glide emodel=-108.324)同实验结构中的配体进行叠合(图 4),二者的RMSD仅为0.7Å,表明上述模型以及对接软件可用于接下来的分子对接研究.
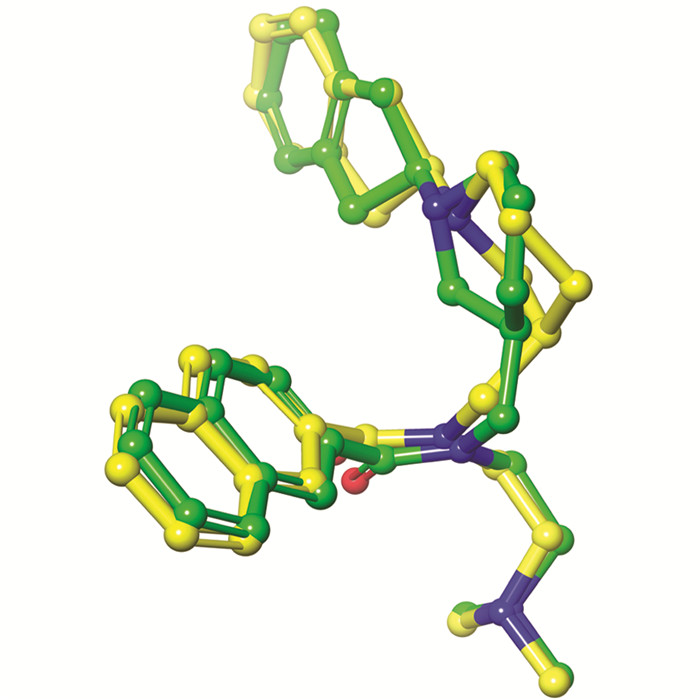
随后选择Glide模块中的SP,XP开展级联分子对接,基于对接精度以及打分函数以不断缩小筛选范围. 首先经普通精度的SP分子对接后,选择表现较好的64个对接构象(Docking score<-25.08 kJ/mol)进行更高精度的XP对接研究. 对表现较好的62个对接构象(XP GScore<-41.8 kJ/mol)经Canvas去重处理后获得4个已上市药物分子结构(图 5):拓扑异构酶II抑制剂米托蒽醌(XP GScore=-57.68 kJ/mol),组蛋白脱乙酰酶抑制剂帕比司他(XP GScore=-51.00 kJ/mol),中枢神经系统兴奋剂赖斯丹明(XP GScore=-44.73 kJ/mol),血管紧张素转化酶抑制剂赖诺普利(XP GScore=-44.73 kJ/mol).

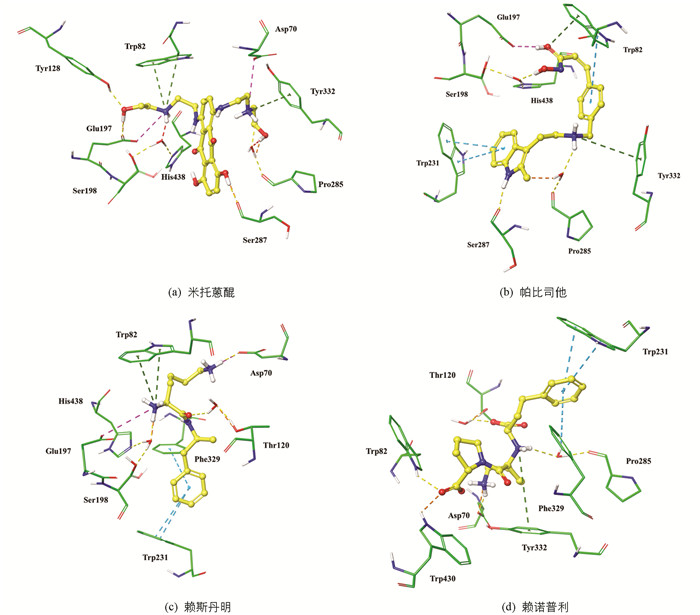
随后,对4个药物分子在BuChE活性位点内的预测结合模式进行分析,图 6a显示米托蒽醌能够同活性位点峡谷中的关键氨基酸Asp70,Trp82,Tyr332产生相互作用,同时通过水分子(HOH805)同催化氨基酸Ser198,His438产生相互作用. 图 6b显示帕比司他同活性位点峡谷中的关键氨基酸Trp82,Trp231,Tyr332产生相互作用,其N-羟基丙烯酰胺侧链通过水分子(HOH805)同催化氨基酸Ser198,His438产生相互作用. 图 6c显示赖斯丹明与蛋白关键氨基酸Asp70,Trp82,Trp231产生相互作用,其结构中的伯氨基通过水分子(HOH805)同催化氨基酸Ser198,His438产生相互作用. 图 6d显示赖诺普利与蛋白关键氨基酸Asp70,Trp82,Trp231,Tyr332产生相互作用,但有别于上述3个药物分子,赖诺普利同催化氨基酸Ser198,His438未产生相互作用.
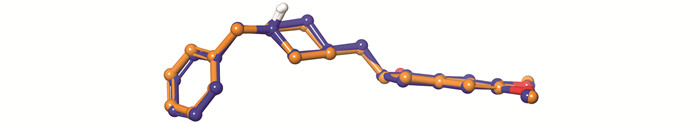
我们对上述4个药物分子就AChE的结合能力进行了预测,对人源AChE与其选择性抑制剂多奈哌齐的复合物晶体结构(PDB ID:4EY7)进行准备后,获得了一个不包含水分子的grid文件,随后通过重对接对grid文件以及对接软件(方法)进行考察. 重对接结果表明,当选择代表性对接构象(Glide emodel=-99.779)同实验结构中的配体进行叠合(图 7),二者的RMSD仅为0.2Å,表明上述模型以及对接软件可用于接下来的分子对接研究.
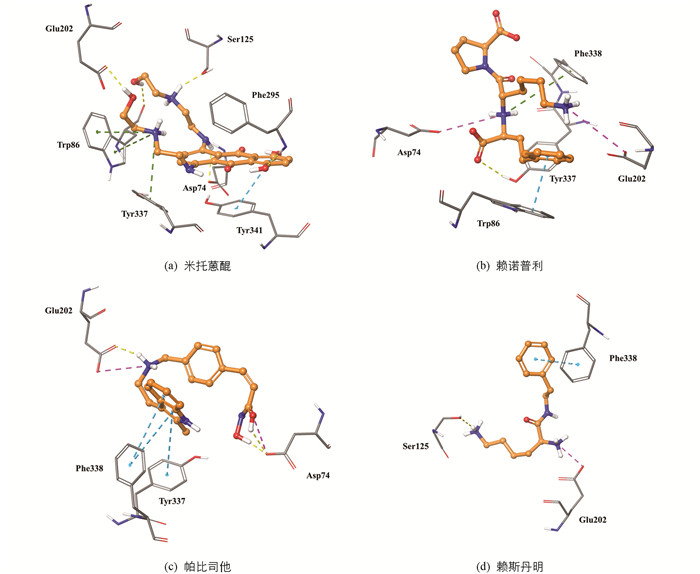
结果显示4个药物分子均对AChE表现出较强的亲和力:米托蒽醌(XP GScore=-66.88 kJ/mol),帕比司他(XP GScore=-72.73 kJ/mol),赖斯丹明(XP GScore=-53.92 kJ/mol),赖诺普利(XP GScore=-51.83 kJ/mol). 对4个药物在AChE活性位点内的预测结合模式进行分析:米托蒽醌与赖诺普利能够同活性位点峡谷中的关键氨基酸Asp74,Trp86,Tyr337等产生相互作用(图 8a,b);帕比司他通过形成U型构象同关键氨基酸Asp74,Trp337产生相互作用(图 8c);赖斯丹明与AChE间未发现关键分子间的相互作用(图 8d).
-
中枢神经系统(CNS)药物的开发面临的众多挑战之一是药物分子能否顺利穿透血脑屏障(BBB). 同时,为了提高药物研发的成功率,医药界也越来越重视在研发早期即对先导化合物的理化性质开展研究. 在本研究中,采用Schrödinger软件中的药动性质(吸收、分布、代谢、排泄)预测程序(QikProp)对4个药物的血脑屏障渗透性进行预测. 表 1显示,除帕比司他未获得预测结果外,其余3个药物均显示具有潜在的BBB渗透性,具备作为CNS药物开发的潜力. 米托蒽醌(QPlogBB=-2.283)的表现不如赖斯丹明(QPlogBB=-0.390)与赖诺普利(QPlogBB=-0.988),我们认为这与其结构中含有多个极性官能团导致其亲水性更强有关.
-
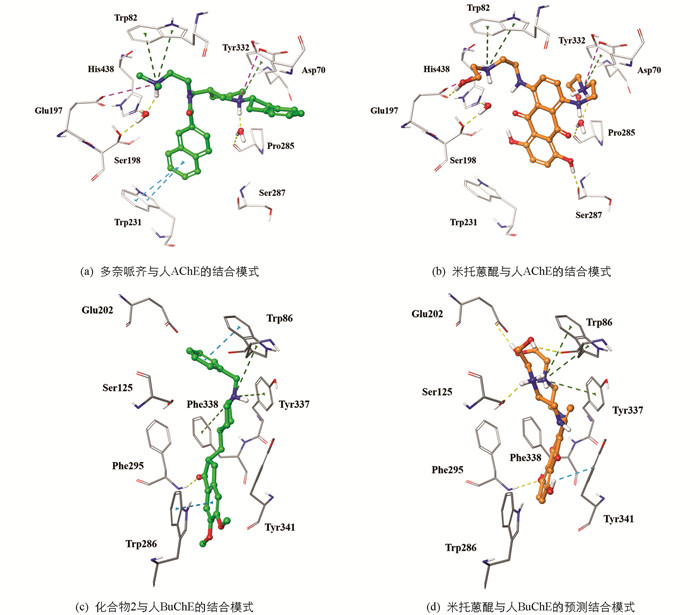
采用优化后的Ellman法,对3个药物(米托蒽醌,帕比司他,赖诺普利)的胆碱酯酶体外抑制活性进行了实际验证,他克林作为阳性对照. 在10 μmol/L测试浓度下,米托蒽醌的抑制率最高(表 2). 米托蒽醌是一类抗肿瘤抗生素,临床用于治疗急性白血病、淋巴瘤以及前列腺癌与乳腺癌,也可用于治疗晚期多发性硬化症. 对其进一步的半数抑制浓度(IC50)研究结果显示:米托蒽醌是一个有效的AChE抑制剂(IC50=0.85 μmol/L)与BuChE抑制剂(IC50=6.45 μmol/L),对AChE的抑制活性优于对BuChE的抑制活性. 将米托蒽醌在胆碱酯酶活性位点内的预测构象与共结晶配体的结合构象进行对比发现,在AChE活性位点内,同共结晶配体多奈哌齐(IC50=7.0 μmol/L)[21]相比(图 9a),米托蒽醌未与关键氨基酸Trp86,Trp286形成π-π堆积作用. 在Phe338附近由于缺乏电荷中心,未能与其形成阳离子-π相互作用(图 9b). 在BuChE活性位点内同共结晶配体化合物2(IC50=0.001 μmol/L,图 9c)相比,米托蒽醌缺乏与关键氨基酸Trp231形成T型π-π堆积作用,并且未能与结晶水(HOH805)形成分子间氢键从而与催化氨基酸His438,Ser198产生联系(图 9d). 上述分析为接下来的结构优化工作提供了思路.
2.1. 配体库的准备与讨论
2.2. 药效团的构建、基于药效团的虚拟筛选与结果分析
2.3. 分子对接及结果分析
2.4. BBB渗透性预测
2.5. 体外胆碱酯酶抑制活性结果及分析
-
AChE是经典的抗AD药物靶标,而随着研究的深入,靶向BuChE对治疗中晚期AD的重要性也越来越得到研究者们的重视. 随着研发投入的不断加大以及开发CNS药物面临的高失败风险,首创药物的创制面临着巨大的挑战. 本研究采用“老药新用”以及“基于结构的虚拟筛选”策略,先根据胆碱酯酶抑制剂的典型结构特征对FDA批准上市药物数据库进行过滤,随后基于药效团模型以及分子对接开展虚拟筛选. 对打分表现较好的药物同胆碱酯酶的预测结合模式进行分析,并对其BBB渗透性进行预测以提高命中率以及合理性. 在体外活性测试中,抗肿瘤药物米托蒽醌被发现具有有效的胆碱酯酶抑制活性,其对AChE的抑制活性优于BuChE抑制活性. 本研究为胆碱酯酶抑制剂提供了新的骨架结构,同时考虑到上市药物的诸多优点,也为接下来高活性、高成药性的胆碱酯酶抑制剂研究提供了理想的研究起点.
 DownLoad:
DownLoad: